
Huisje, tuintje, … : woonmodel met toekomstpotentieel?
Het Vlaamse woonmodel staat voor een grote uitdaging in het licht van de veelbesproken ‘betonstop’: tegen 2040 moet het bijkomend ruimtebeslag naar 0, hoewel het aantal huishoudens erg zal stijgen. Is grondgebonden wonen nog mogelijk en kan het aanzien worden als een duurzame woonvorm om de Antwerpse stadsregio te verdichten zonder bijkomend ruimtebeslag? Deze studie analyseert de bestaande woonsituatie en verdichtingsprincipes om via concreet ontwerpend onderzoek de haalbaarheid ervan af te toetsen aan de noden van de toekomst. Tot slot geeft dit werk een aanzet tot de uitwerking van een draagkrachtige beleidsvisie.
Het actueel ruimtelijk beleid van de Vlaamse regering zet in op de verhoging van het ruimtelijk rendement in de stedelijke gebieden. Cijfers tonen aan dat de toename van het aantal huishoudens voor de stadsregio Antwerpen beduidend lager is dan het Vlaamse gemiddelde en de actualisering van deze prognose uit 2017 is zelfs nog pessimistischer. Het is zorgwekkend dat, ondanks de reeds geleverde inspanningen, suburbanisatie nog steeds verder gaat.
Om suburbanisatie te stoppen, is er in de Antwerpse stadsregio nood aan minimaal 33.337 bijkomende woningen tegen 2027. Concreet staan er in deze regio 116 projecten in planning voor de periode 2014 tot 2024 voor ongeveer 20.000 bijkomende woningen. Deze woonprojecten bestaan hoofdzakelijk uit appartementen die nog te vaak in open ruimte op een grote afstand van het bestaande woonweefsel worden gepland. De inplanting in het bestaande woonweefsel wordt vermeden uit vrees voor weerstand van de omwonenden. Veel van deze projecten zijn bovendien onvoldoende afgestemd op het bestaande openbaarvervoernetwerk.
Het pad vrijmaken om de revolutionaire visie in praktijk te brengen zoals de ontwikkeling van woontorens in de dorpskernen, zal enorm veel tijd kosten. Kostbare tijd die er niet is als je ziet hoeveel nieuwe ruimte er dagelijks wordt ingenomen. In de stadsregio worden momenteel aan een sneltempo grootschalige appartementiseringsprojecten gerealiseerd. Deze trend duwt jonge gezinnen weg uit de stedelijke omgeving en stimuleert suburbanisatie. Het radicaal overboord gooien van de Vlaamse bouwcultuur kan nefaste gevolgen hebben voor de stadsrand, die de taakstelling van de stad deels moet overnemen. Dit onderzoek toont aan dat het ook anders kan. Ook volgens de bestaande Vlaamse bouwcultuur en zonder bijkomend ruimtebeslag kan er naar een duurzame oplossing worden gezocht. Op korte termijn inzetten op het verzoenen van de Vlaamse woonvoorkeur, een rustig gelegen woning met tuin, en duurzame ruimtelijke planning kan de gezinnen motiveren om in de stadsregio te blijven.
Duurzaam ruimtegebruik kan enkel bekomen worden door dichter bij elkaar te wonen in goed ontsloten leefbare kernen mits bewaking van de ‘pretfactor’ die volgens de Vlaams Bouwmeester aan de woonlocatie moet verbonden zijn. Met de toepassing van kleinschalige grondgebonden verdichting wordt gewerkt aan een draagvlak voor stedelijke inbreidingsprojecten in de stadsrand. De verstedelijking van de dorpen in de stadsrand is onvermijdelijk maar moet gebeuren met respect voor het bestaande ‘dorpse’ karakter.
Het realiseren van kleinschalige particuliere BIMBY (Build In My BackYard) projecten waarbij in de tuin een bescheiden of volwaardige woonunit wordt gebouwd op de plaats, waar vaak een tuinhuis of een garage staat, kan tegemoetkomen aan de woonbehoefte en woonvoorkeur. Kleinschalige ‘tuingroep’ projecten, waarbij enkele woningen een tuin delen vormen eveneens een mogelijkheid. Als deze verdichting enkel en alleen gerealiseerd wordt op de met openbaar vervoer bereikbare percelen binnen het bestaande woonweefsel van de halfopen en open gezinswoningen, kan het noodzakelijke aantal bijkomende woningen gehaald worden.

Voor verdichting

Na toevoeging van 19 woningen
De concentratie van verdichting in bestaande duurzame buurten op slimme locaties levert, naast de verhoging van het ruimtelijk rendement, ook andere voordelen op. Het meer diverse woonaanbod versterkt de sociale cohesie. Het groter aantal bewoners, ofwel een toename van de kritische massa zorgt ervoor dat op deze strategische locaties handelskernen voor lokale economie opnieuw kansen krijgen. Naast de maatschappelijke meerwaarde biedt het concept financiële voordelen voor zowel de koper als verkoper zodat de winsten rechtstreeks bij de burger terechtkomen. Bovendien groeit op deze manier het maatschappelijk besef van de individuele burger om op een verantwoorde manier met de aarde om te gaan. Kortom, het concept slaagt erin om private en collectieve belangen te verzoenen.
Het onderzoek toont voor het grootstedelijk gebied Antwerpen aan dat grondgebonden wonen kan ingezet worden als een duurzame verdichtingsvorm. Het concreet ontwerpend onderzoek is opgebouwd uit een kwantitatief onderzoek waarbij op basis van drie scenario’s het bijkomend aantal woningen wordt berekend. Twee scenario’s zijn gebaseerd op de doelstelling uit het Beleidsplan Ruimte Vlaanderen. Het derde past de internationale verdichtingstheorie ‘Transit Oriented Development’ toe waarbij gestreefd wordt naar hoge woondichtheden binnen het bereik van openbaar vervoer. De eerste twee scenario’s blijken weinig ambitieuze resultaten op te leveren terwijl het derde scenario een ingrijpende transformatie nastreeft. Dit kwantitatieve onderzoek heeft als doel het lokaal beleid vorm te geven en de resultaten in te schatten. Als voorbeeld werd er voor het concrete projectgebied in de gemeente Hemiksem een kwalitatief ontwerp opgebouwd uit drie stappen. De eerste stap is een ‘reconfiguration’ strategie waarbij enkel op basis van kleinschalige initiatieven door private bouwheren verdicht wordt (geel). De tweede stap is de ‘replacement’ strategie waarbij meer grootschalige herzieningen van de kavelstructuren doorgevoerd worden naar kleinschalige meergezinswoningprojecten of inbreidingsprojecten in binnengebieden (oranje). De derde stap is de grootschaligere ‘removal’ strategie waarbij via sloop van het bestaande patrimonium plaats gemaakt wordt voor nieuwe ontwikkelingen (rood).
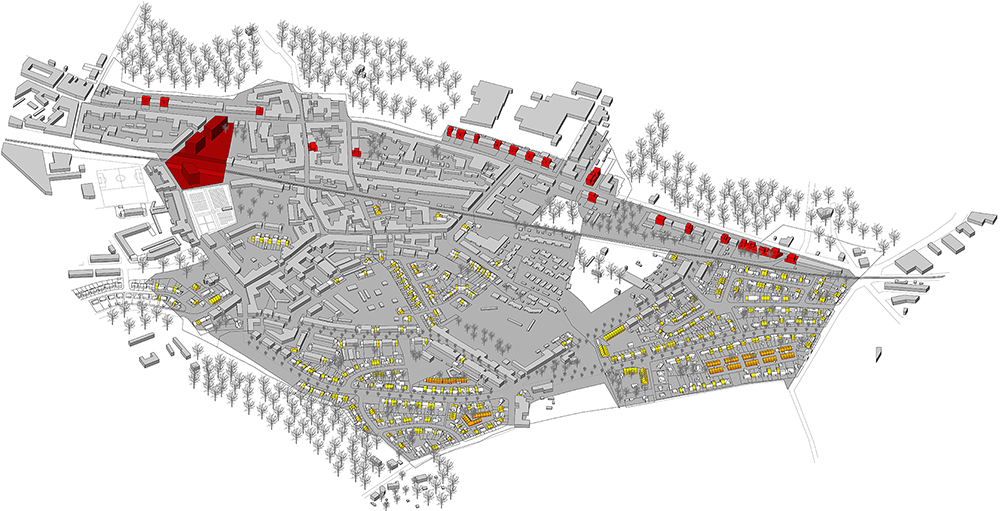
Eigen model – voorstel stap 3: Removal
Om deze verdichtingsstrategie om te zetten naar de praktijk is er nood aan een draagkrachtige beleidsvisie. De bevolking en de private bouwheren zullen hierbij een cruciale rol spelen. Zij kunnen ofwel positief meewerken en het beleid vormgeven ofwel met een NIMBY (Not In My BackYard) houding een trend naar verdichting bevriezen. Daarbij is er nood aan ruimtelijke sturing die onvoldoende kwalitatieve ontwikkelingen op ongewenste locaties radicaal een halt toeroept. De recente beleidsvisies leggen de verantwoordelijkheid hiervoor bij de lokale besturen omdat zij het best vertrouwd zijn met de lokale processen. Dat klopt in realiteit niet. Het realiseren van de ruimtebehoefte zonder bijkomend ruimtebeslag is een bovenlokaal proces waarvoor een concreet duurzaam planningskader noodzakelijk is. De opmaak van dit planningskader moet dus getrokken worden door de Vlaamse Overheid met input van de lokale kennis.







