Superbacteriën bestrijden met behulp van slimme computertechnieken
Stel u voor: we bevinden ons in 2050. Bijna alle bacteriën zijn nu resistent geworden aan de antibiotica waarmee we ze ooit eenvoudig konden bestrijden. Sterker nog, jaarlijks gaan nu enkele miljoenen mensen per jaar dood aan allerhande infecties die vroeger eenvoudig te behandelen waren. We zijn teruggekeerd naar het pre-antibiotica tijdperk, waar nu bacteriële infecties door zogenaamde ‘superbacteriën’ een van de belangrijkste doodsoorzaken zijn geworden. Gelukkig genoeg zijn we nog niet in 2050, en kunnen we dit scenario vermijden. En wel door virussen te gebruiken om bacteriën te bestrijden. Virussen, hoor ik u denken?!
Virussen zijn de kleinste en meest diverse biologische entiteiten op onze planeet. Alle virussen hebben een ander organisme nodig, de gastheer, om zichzelf te kunnen voortplanten. Sommige virussen gebruiken bacteriën als gastheer. Zo’n virussen noemen we fagen. Het zijn zeer kleine, eenvoudige virussen die bacteriën infecteren, en na infectie hun gastheer ook kunnen afdoden. Inderdaad, in de laatste stap van hun levenscyclus gaan vele fagen hun bacteriële gastheer afdoden zodat zijzelf opnieuw kunnen verspreiden naar nieuwe gastheren. Dit maakt hen interessant als alternatief bestrijdingsmiddel tegen bacteriën. Daarnaast heeft het gebruik van fagen twee voordelen in vergelijking met klassieke antibiotica: fagen zijn specifiek en ze evolueren. Vele klassieke antibiotica zijn niet specifiek (genoeg), waardoor ze soms nefaste effecten hebben op de nuttige bacteriën in ons lichaam. Fagen zijn daarentegen zeer specifiek, ze kunnen vaak slechts één bepaalde soort bacterie of enkele sterk gerelateerde soorten infecteren. Daarnaast evolueren fagen ook mee met bacteriën (een proces dat co-evolutie wordt genoemd), waardoor ze opnieuw de bovenhand kunnen nemen als hun bacteriële gastheer resistent wordt.
Fagen zijn kieskeurig
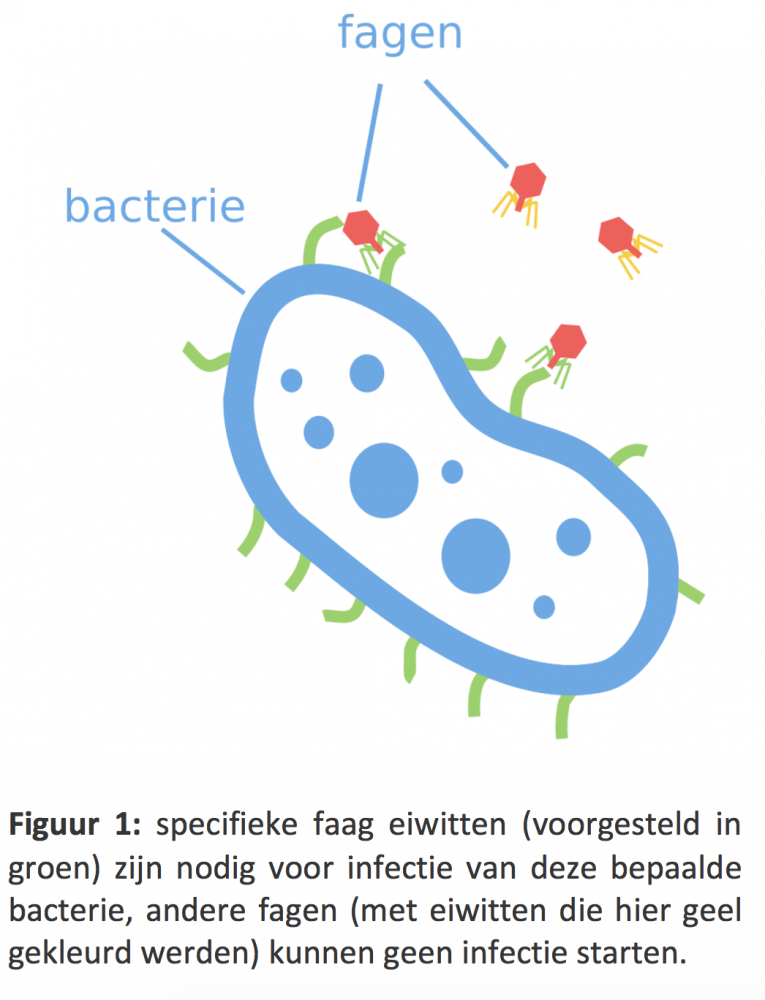
Fagen weten goed wat ze willen: ze kiezen bijna altijd resoluut voor het infecteren van één specifieke soort bacterie of sterk gerelateerde soorten. Fagen vertonen deze specificiteit ten opzichte van hun bacteriële gastheren doordat ze gebruik maken van specifieke eiwitten die geschikte gastheren herkennen. Figuur 1 geeft een illustratie van hoe deze specifieke eiwitten bepaalde componenten van de bacterie selectief kunnen herkennen. Om fagen te kunnen gebruiken als bestrijdingsmiddel tegen bacteriën is het belangrijk om een goed begrip te hebben van de specificiteit van fagen. Aangezien specifieke faag eiwitten hier de belangrijkste factor in zijn, stelde mijn thesis de volgende vraag: kunnen we de specificiteit van fagen begrijpen en identificeren op het niveau van faag eiwitten? Om deze vraag te beantwoorden ging ik niet naar het laboratorium, maar kroop ik achter mijn computer.
Computers gebruiken als hefboom
De specificiteit van fagen bestuderen kan op verschillende manieren. De meest gebruikte manier is door het uitvoeren van een eenvoudige test in het laboratorium: wetenschappers zetten fagen bij de bacterie waarin ze geïnteresseerd zijn en nemen dan waar of de groei van die bacterie afneemt door aanwezigheid van de faag. Er zijn echter zo veel fagen dat het onbegonnen werk is om ze allemaal te testen in het laboratorium. Zo’n tests zijn ook minder geschikt voor het ontdekken van complexere patronen in interacties, bv. tussen fagen die dezelfde soort bacterie infecteren. Hier kunnen computers echter bij helpen. De hypothese is dat computers veel sneller grote hoeveelheden data kunnen analyseren, om zo patronen te ontdekken die nooit duidelijk zouden worden door het één voor één bestuderen van interacties. In mijn thesis heb ik daarom twee computertechnieken gebruikt om data van fagen te analyseren: optimal transport en machine learning.
Optimal transport is een nieuwe manier van vergelijken
Optimal transport is een wiskundige techniek die toelaat om (wiskundig geformuleerde) objecten te vergelijken. Op die manier kan de techniek gebruikt worden om afstanden te berekenen tussen deze objecten. Ik heb aldus fagen (en meer specifiek de verzameling van hun eiwitten) wiskundig voorgesteld en vergeleken via optimal transport. De voornaamste doelen waren om aan te tonen dat optimal transport gebruikt kan worden om biologische data te bestuderen, en om eiwitten te vinden die uniek waren tussen gelijkaardige fagen die toch een andere bacteriële gastheer hebben. Deze unieke eiwitten kunnen dan (deels) het verschil in gastheer specificiteit verklaren.
Machine learning en biologie zijn een goede combinatie
De eiwitten die fagen gebruiken om hun gastheer te herkennen zijn vaak gelegen op de staarten van deze fagen. Deze staart eiwitten (in het Engels ‘tail fiber’ en ‘tail spike’ eiwitten genaamd) zijn vaak de primaire determinanten van gastheer specificiteit. Daarom werden deze eiwitten in het laatste deel van mijn thesis gebruikt om de bacteriële gastheer te voorspellen. Hiervoor werd machine learning gebruikt. Machine learning is een wetenschappelijke discipline die algoritmen ontwikkelt die patronen ontdekken in data. Zo kan de computer ‘uit zichzelf’ leren hoe bepaalde data eruitzien en hier regels aan koppelen. Ik heb machine learning gebruikt om regels te ontdekken in verband met de bacteriële gastheer van fagen, door data van de staart eiwitten van die fagen aan ‘slimme’ algoritmen mee te geven. Het resultaat was verassend goed. De computer is in staat om tot in 93% van de gevallen de juiste gastheer te voorspellen op basis van het staart eiwit van de faag. Aangezien het bepalen van de gastheer van een faag in het laboratorium niet altijd eenvoudig is, kunnen deze algoritmen zeker een meerwaarde bieden in het voorspellen van de gastheer. Dit is namelijk een cruciale stap in het gebruik van fagen als alternatief bestrijdingsmiddel tegen bacteriën.
Wat betekent dit voor u?
In mijn thesis heb ik de specificiteit van bacterie-faag interacties bestudeerd via computertechnieken. De tools die ik heb ontwikkeld kunnen mee de basis helpen vormen van een alternatieve strategie om bacteriën te bestrijden, zodat we niet langer enkel afhankelijk zijn van klassieke antibiotica waartegen snel resistentie optreedt. Concreet betekent dit dat u op uw twee oren kan slapen. De wetenschap staat nooit stil en zal altijd oplossingen blijven zoeken (en vinden) voor de problemen van vandaag en morgen!
Bibliografie
Ackermann, H. W. (2007). 5500 Phages examined in the electron microscope. Archives of Virology, 152(2), 227243. http://doi.org/10.1007/s00705-006-0849-1
Adriaenssens, E. M., Edwards, R., Nash, J. H. E., Mahadevan, P., Seto, D., Ackermann, H. W., Kropinski, A. M. (2015). Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology, 477, 144154. http://doi.org/10.1016/j.virol.2014.10.016
Ahmed, S., Saito, A., Suzuki, M., Nemoto, N., and Nishigaki, K. (2009). Host-parasite relations of bacteria and phages can be unveiled by Oligostickiness, a measure of relaxed sequence similarity. Bioinformatics, 25(5), 563570. http://doi.org/10.1093/bioinformatics/btp003
Al-Shahib, A., Breitling, R., and Gilbert, D. R. (2007). Predicting protein function by machine learning on amino acid sequences - A critical evaluation. BMC Genomics, 8. http://doi.org/10.1186/1471-2164-8-78
Ando, H., Lemire, S., Pires, D. P., and Lu, T. K. (2015). Engineering Modular Vi- ral Scaffolds for Targeted Bacterial Population Editing. Cell Systems, 1(3), 187196. http://doi.org/10.1016/j.cels.2015.08.013
Bateman, A., Martin, M. J., ODonovan, C., Magrane, M., Alpi, E., Antunes, R., Zhang, J. (2017). UniProt: The universal protein knowledgebase. Nucleic Acids Research, 45(D1), D158D169. http://doi.org/10.1093/nar/gkw1099
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2013). GenBank. Nucleic Acids Research, 41(D1), 3642. http://doi.org/10.1093/nar/gks1195
Cenens, W., Makumi, A., Mebrhatu, M. T., Lavigne, R., and Aertsen, A. (2013). Phage-host interactions during pseudolysogeny. Bacteriophage, 3(1), e25029. http://doi.org/10.4161/bact.25029
Cerritelli, M. E., Wall, J. S., Simon, M. N., Conway, J. F., and Steven, A. C. (1996). Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: A hinged viral adhesin. Journal of Molecular Biology, 260(5), 767780. http://doi.org/10.1006/jmbi.1996.0436
Chaturongakul, S., and Ounjai, P. (2014). Phage-host interplay: Examples from tailed phages and Gram-negative bacterial pathogens. Frontiers in Microbiology, 5(AUG), 18. http://doi.org/10.3389/fmicb.2014.00442
Clokie, M. R. J., Millard, A. D., Letarov, A. V., and Heaphy, S. (2011). Phages in nature. Bacteriophage, 1(1), 3145. http://doi.org/10.4161/bact.1.1.14942
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., De Hoon, M. J. L. (2009). Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 14221423. http://doi.org/10.1093/bioinformatics/btp163
Cuervo, A., Pulido-Cid, M., Chagoyen, M., Arranz, R., González-García, V. A., Garcia- Doval, C., Carrascosa, J. L. (2013). Structural characterization of the bacteriophage T7 tail machinery. Journal of Biological Chemistry, 288(36), 2629026299. http://doi.org/10.1074/jbc.M113.491209
Cui, J., Yang, B. (2018). Graph Bayesian Optimization: Algorithms, Evaluations and Applications. Journal of Machine Learning Research, 18, 1-24.
Cuturi, M. (2013). Sinkhorn Distances: Lightspeed Computation of Optimal Transportation Distances, 113. Retrieved from http://arxiv.org/abs/1306.0895
Das, S., Deb, T., Dey, N., Ashour, A. S., Bhattacharya, D. K., and Tibarewala, D. N. (2017). Optimal choice of k-mer in composition vector method for genome sequence comparison. Genomics, (November), 01. http://doi.org/10.1016/j.ygeno.2017.11.003
Daudén, M. I., Marti-Benito, J., Sánchez-Ferrero, J. C., Pulido-Cid, M., Valpuesta, J. M., and Carrascosa, J. L. (2013). Large terminase conformational change induced by connector binding in bacteriophage T7. Journal of Biological Chemistry, 288(23), 1699817007. http://doi.org/10.1074/jbc.M112.448951
Davies, J., and Davies, D. (2010). Origins and Evolution of Antibiotic Resistance. Microbiology and Molecular Biology Reviews, 74(3), 417433. http://doi.org/10.1128/MMBR.00016-10
Deaton, J., Yu, F. B., and Quake, S. R. (2017). PhaMers identifies novel bacteriophage sequences from thermophilic hot springs, (September), 131. http://doi.org/10.1101/126847
Díaz-Muñoz, S. L., and Koskella, B. (2014). Bacteria-Phage interactions in natural environments. Advances in Applied Microbiology (1st ed., Vol. 89). Elsevier Inc. http://doi.org/10.1016/B978-0-12-800259-9.00004-4
Doss, J., Culbertson, K., Hahn, D., Camacho, J., and Barekzi, N. (2017). A review of phage therapy against bacterial pathogens of aquatic and terrestrial organisms. Viruses, 9(3). http://doi.org/10.3390/v9030050
Duplessis, M., and Moineau, S. (2001). Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Molecular Microbiology, 41(2), 325336. http://doi.org/10.1046/j.1365-2958.2001.02521.x
Dupont, K., Vogensen, F. K., Neve, H., Bresciani, J., and Josephsen, J. (2004). Identification of the receptor-binding protein in 936-species lactococcal bacteriophages. Applied and Environmental Microbiology, 70(10), 58185824. http://doi.org/10.1128/AEM.70.10.5818-5824.2004
Edwards, R. A., McNair, K., Faust, K., Raes, J., and Dutilh, B. E. (2016). Computational approaches to predict bacteriophage-host relationships. FEMS Microbiology Reviews, 40(2), 258272. http://doi.org/10.1093/femsre/fuv048
Egley, L. E. W., and Breitbart, M. (2003). Use of Fluorescently Labeled Phage in the Detection and Identification of Bacterial Species. Applied Spectroscopy, 57(9), 7. http://doi.org/10.1366/00037020360696008
Eickholt, J., and Wang, Z. (2014). PCP-ML: Protein characterization package for Machine Learning. BMC Research Notes, 7(1), 15. http://doi.org/10.1186/1756-0500-7- 810
Fàbrega, A., and Vila, J. (2013). Salmonella enterica serovar Typhimurium skills to succeed in the host: Virulence and regulation. Clinical Microbiology Reviews, 26(2), 308341. http://doi.org/10.1128/CMR.00066-12
Fineran, P. C., Blower, T. R., Foulds, I. J., Humphreys, D. P., Lilley, K. S., and Salmond, G. P. C. (2009). The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proceedings of the National Academy of Sciences, 106(3), 894899. http://doi.org/10.1073/pnas.0808832106
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., Mitchell, A. L. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Research, 45(D1), D190D199. http://doi.org/10.1093/nar/gkw1107
Fokine, A., and Rossmann, M. G. (2014). Molecular architecture of tailed double-stranded DNA phages. Bacteriophage, 4(2), e28281. http://doi.org/10.4161/bact.28281
Ghequire, M. G. K., and De Mot, R. (2015). The Tailocin Tale: Peeling off Phage Tails. Trends in Microbiology, 23(10), 587590. http://doi.org/10.1016/j.tim.2015.07.011
Giske, C. G., Monnet, D. L., Cars, O., and Carmeli, Y. (2008). Clinical and economic impact of common multidrug-resistant gram-negative bacilli. Antimicrobial Agents and Chemotherapy, 52(3), 813821. http://doi.org/10.1128/AAC.01169-07
Guruprasad, K., Reddy, B. V. B., and Pandit, M. W. (1990). Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Engineering, Design and Selection, 4(2), 155161. http://doi.org/10.1093/protein/4.2.155
Hobohm, U., and Sander, C. (1995). A sequence property approach to searching protein databases. Journal of Molecular Biology, 251(3), 390399. http://doi.org/10.1006/jmbi.1995.0442
Hoess, R. H., and Landy, A. (1978). Structure of the lambda att sites generated by int-dependent deletions. Proceedings of the National Academy of Sciences, 75(11), 54375441. http://doi.org/10.1073/pnas.75.11.5437
Horvath, P., Barrangou, R. (2010). CRISPR/Cas, the Immune System of Bacteria and Archaea. Source: Science, New Series, 327(5962), 167170. http://doi.org/10.1126/science.1179555
Hyman, P., and Abedon, S. T. (2010). Bacteriophage host range and bacterial resistance. Advances in applied microbiology (1st ed., Vol. 70). Elsevier Inc. http://doi.org/10.1016/S0065-2164(10)70007-1
Jiang, S. C., and Paul, J. H. (1996). Occurrence of lysogenic bacteria in marine microbial communities as determined by prophage induction. Marine Ecology Progress Series, 142, 2738. http://doi.org/10.3354/meps142027
Kiro, R., Molshanski-Mor, S., Yosef, I., Milam, S. L., Erickson, H. P., and Qimron, U. (2013). Gene product 0.4 increases bacteriophage T7 competitiveness by inhibiting host cell division. Proceedings of the National Academy of Sciences, 110(48), 1954919554. http://doi.org/10.1073/pnas.1314096110
Koskella, B., and Meaden, S. (2013). Understanding bacteriophage specificity in nat- ural microbial communities. Viruses, 5(3), 806823. http://doi.org/10.3390/v5030806
Kwan, T., Liu, J., DuBow, M., Gros, P., and Pelletier, J. (2005). The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proceedings of the National Academy of Sciences, 102(14), 51745179. http://doi.org/10.1073/pnas.0501140102
Latka, A., Maciejewska, B., Majkowska-Skrobek, G., Briers, Y., and Drulis-Kawa, Z. (2017). Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Applied Microbiology and Biotechnology, 101(8), 31033119. http://doi.org/10.1007/s00253-017-8224-6
Lavigne, R., Seto, D., Mahadevan, P., Ackermann, H. W., and Kropinski, A. M. (2008). Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Research in Microbiology, 159(5), 406414. http://doi.org/10.1016/j.resmic.2008.03.005
Le, S., He, X., Tan, Y., Huang, G., Zhang, L., Lux, R., Hu, F. (2013). Mapping the Tail Fiber as the Receptor Binding Protein Responsible for Differential Host Specificity of Pseudomonas aeruginosa Bacteriophages PaP1 and JG004. PLoS ONE, 8(7), 18. http://doi.org/10.1371/journal.pone.0068562
Leite, D. M. C., Brochet, X., Resch, G., Que, Y.-A., Neves, A., and Pena-Reyes, C. (2017). Computational Prediction of Host-Pathogen Interactions Through Omics Data Analysis and Machine Learning, 10209, 1530. http://doi.org/10.1007/978-3-319-56154-7
Lenski, R. E. (1984). Coevolution of bacteria and phage: Are there endless cycles of bacterial defenses and phage counterdefenses? Journal of Theoretical Biology, 108(3), 319325. http://doi.org/10.1016/S0022-5193(84)80035-1
Lévy, B., and Schwindt, E. L. (2018). Notions of optimal transport theory and how to implement them on a computer. Computers and Graphics (Pergamon), 72, 135148. http://doi.org/10.1016/j.cag.2018.01.009
Lindberg, A. A. (1973). Bacteriophage Receptors.
Lipman, D. J., Wilbur, W. J., Smith, T. F., and Waterman, M. S. (1984). On the statistical significance of nucleic acid similarities. Nucleic Acids Research, 12(1 Pt 1), 215226. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC320998/
Lobry, J. R., and Gautier, C. (1994). Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 escherichia coli chromosome-encoded genes. Nucleic Acids Research, 22(15), 31743180. http://doi.org/10.1093/nar/22.15.3174
Mahmood, K., Webb, G. I., Song, J., Whisstock, J. C., and Konagurthu, A. S. (2012). Efficient large-scale protein sequence comparison and gene matching to identify orthologs and co-orthologs. Nucleic Acids Research, 40(6). http://doi.org/10.1093/nar/gkr1261
Maxwell, K. L., and Frappier, L. (2007). Viral Proteomics. Microbiology and Molecular Biology Reviews, 71(2), 398411. http://doi.org/10.1128/MMBR.00042-06
Mendelman, L. V, Notarnicola, S. M., and Richardson, C. C. (1992). Roles of bac- teriophage T7 gene 4 proteins in providing primase and helicase functions in vivo. Proceedings of the National Academy of Sciences of the United States of America, 89(22), 1063842. http://doi.org/10.1073/pnas.89.22.10638
Menze, B. H., Kelm, B. M., Masuch, R., Himmelreich, U., Bachert, P., Petrich, W., and Hamprecht, F. A. (2009). A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinformatics, 10, 116. http://doi.org/10.1186/1471-2105-10-213
Middelboe, M., Chan, A. M., and Bertelsen, S. K. (2010). Isolation and life cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria. Manual of Aquatic Viral Ecology, (May 2018), 118133. http://doi.org/10.4319/mave.2010.978- 0-9845591-0-7.118
Mihara, T., Nishimura, Y., Shimizu, Y., Nishiyama, H., Yoshikawa, G., Uehara, H., Ogata, H. (2016). Linking virus genomes with host taxonomy. Viruses, 8(3), 1015. http://doi.org/10.3390/v8030066
Modi, S. R., Lee, H. H., Spina, C. S., and Collins, J. J. (2013). Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature, 499(7457), 219222. http://doi.org/10.1038/nature12212
Mokili, J. L., Rohwer, F., and Dutilh, B. E. (2012). Metagenomics and future perspectives in virus discovery. Current Opinion in Virology, 2(1), 6377. http://doi.org/10.1016/j.coviro.2011.12.004
Moldovan, R., Chapman-McQuiston, E., and Wu, X. L. (2007). On kinetics of phage adsorption. Biophysical Journal, 93(1), 303315. http://doi.org/10.1529/biophysj.106.102962
Monge, G. (1781). Mémoire sur la théorie des déblais et des remblais. Histoire de lAcadmie Royale des Sciences (1781), 666704, 1784. (Original reference unatainable, cited from Lévy and Schwindt, 2017).
Nishimura, Y., Yoshida, T., Kuronishi, M., Uehara, H., Ogata, H., and Goto, S. (2017). ViPTree: The viral proteomic tree server. Bioinformatics, 33(15), 23792380. http://doi.org/10.1093/bioinformatics/btx157
ONeill, J. (2016). Tackling drug-resistant infections globally: final report and recom- mendations. The Review on Antimicrobial Resistance, (May), 84. http://doi.org/10.1016/j.jpha.2015.11.005
Park, M., Lee, J. H., Shin, H., Kim, M., Choi, J., Kang, D. H., Ryu, S. (2012). Charac- terization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Applied and Environmental Microbiology, 78(1), 5869. http://doi.org/10.1128/AEM.06231-11
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., Duchesnay, É. (2012). Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research, 12, 28252830. http://doi.org/10.1007/s13398-014-0173-7.2
Pride, D. T., Wassenaar, T. M., Ghose, C., and Blaser, M. J. (2006). Evidence of host-virus co-evolution in tetranucleotide usage patterns of bacteriophages and eukaryotic viruses. BMC Genomics, 7, 113. http://doi.org/10.1186/1471-2164-7-8
Rakhuba, D. V., Kolomiets, E. I., Szwajcer Dey, E., and Novik, G. I. (2010). Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Polish Journal of Microbiology, 59(3), 145155. http://doi.org/10.1016/j.micres.2015.01.008.1.94
Ren, J., Ahlgren, N. A., Lu, Y. Y., Fuhrman, J. A., and Sun, F. (2017). VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome, 5(1), 69. http://doi.org/10.1186/s40168-017-0283-5
Rohwer, F., and Edwards, R. (2002). The Phage Proteomic Tree : a Genome-Based Taxonomy for Phage, 184(16), 45294535. http://doi.org/10.1128/JB.184.16.4529
Rohwer, F., Prangishvili, D., and Lindell, D. (2009). Roles of viruses in the environment. Environmental Microbiology, 11(11), 27712774. http://doi.org/10.1111/j.1462- 2920.2009.02101.x
Roock, S. De, and Steven, M. (2014). A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens, 5(1), 110.
Ross, A., Ward, S., and Hyman, P. (2016). More is better: Selecting for broad host range bacteriophages. Frontiers in Microbiology, 7(SEP), 16. http://doi.org/10.3389/fmicb.2016.01352
Rossolini, G. M., Arena, F., Pecile, P., and Pollini, S. (2014). Update on the antibiotic resistance crisis. Current Opinion in Pharmacology, 18, 5660. http://doi.org/10.1016/j.coph.2014.09.006
Roux, S., Hallam, S. J., Woyke, T., and Sullivan, M. B. (2015). Viral dark matter and virus host interactions resolved from publicly available microbial genomes. eLife, 4(January), 120. http://doi.org/10.7554/eLife.08490
Saitou, N., and Nei, M. (1998). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4(4), 406425. Retrieved from https://academic.oup.com/mbe/article/4/4/406/1029664
Samson, J. E., Magadán, A. H., Sabri, M., and Moineau, S. (2013). Revenge of the phages: Defeating bacterial defences. Nature Reviews Microbiology, 11(10), 675687. http://doi.org/10.1038/nrmicro3096
Sastry, A., Monk, J., Tegel, H., Uhlen, M., Palsson, B. O., Rockberg, J., and Brunk, E. (2017). Machine learning in computational biology to accelerate high-throughput protein expression. Bioinformatics. http://doi.org/10.1093/bioinformatics/btx207
Schmelcher, M., Donovan, D. M., and Loessner, M. J. (2012). Bacteriophage endolysins as novel antimicrobials. Future Microbiology, 7(10), 11471171. http://doi.org/10.2217/fmb.12.97
Scholl, D., Kieleczawa, J., Kemp, P., Rush, J., Richardson, C. C., Merril, C., Molineux, I. J. (2004). Genomic Analysis of Bacteriophages SP6 and K1-5, an Estranged Subgroup of the T7 Supergroup. Journal of Molecular Biology, 335(5), 11511171. http://doi.org/10.1016/j.jmb.2003.11.035
Simmonds, P., Adams, M. J., Benk, M., Breitbart, M., Brister, J. R., Carstens, E. B., Zerbini, F. M. (2017). Consensus statement: Virus taxonomy in the age of metagenomics. Nature Reviews Microbiology, 15(3), 161168. http://doi.org/10.1038/nrmicro.2016.177
Simon, E. J., Reece, J. B., Dickey, J. (2010). Campbell Essential Biology: Fourth Edition. Benjamin Cummings. ISBN 0321772601.
Stern, A., Mick, E., Tirosh, I., Sagy, O., and Sorek, R. (2012). CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Research, 22(10), 19851994. http://doi.org/10.1101/gr.138297.112
Steven, A. C., Trus, B. L., Maize, J. V, Unser, M., Parry, D. A. D., Wal, J. S., Studier, F. W. (1988). Molecular substructure of a viral receptor recognition protein The gp17 tail fiber of bacteriophage T7, 0, 351365.
Studier, F. W. (1975). Gene 0.3 of bacteriophage T7 acts to overcome the DNA restriction system of the host. Journal of Molecular Biology, 94(2), 283295. http://doi.org/10.1016/0022-2836(75)90083-2
Susskind, M. M., and Botstein, D. (1980). Superinfection exclusion by prophage in lysogens of Salmonella typhimurium. Virology, 100(1), 212216. http://doi.org/10.1016/0042-6822(80)90571-1
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B., and Wu, C. H. (2015). UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics, 31(6), 926932. http://doi.org/10.1093/bioinformatics/btu739
Tabor, S., and Richardson, C. C. (1989). Selective inactivation of the exonuclease activity of bacteriophage T7 DNA polymerase by in vitro mutagenesis. Journal of Biological Chemistry, 264(11), 64476458.
Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2703498
Tortora, G. J., Funke, B. R., and Case, C. L. (2013). Microbiology: an introduction, Pearson, Boston, 11th edition. ISBN 9780321733603 (student ed.).
Trevors, J. T. (1999). Evolution of gene transfer in bacteria [Review]. World J Microbiol Biotechnol, 15(1), 17. http://doi.org/10.1023/A:1008830914223
Vihinen, M., Torkkila, E., and Riikonen, P. (1994). Accuracy of protein flexibility predic- tions. Proteins: Structure, Function, and Bioinformatics, 19(2), 141149. http://doi.org/10.1002/prot.340190207
Vinga, S., and Almeida, J. (2003). Alignment-free sequence comparison - A review. Bioinformatics, 19(4), 513523. http://doi.org/10.1093/bioinformatics/btg005
Weigele, P. R., Scanlon, E., and King, J. (2003). Homotrimeric, beta-stranded viral adhesins and tail proteins. Journal of Bacteriology, 185(14), 40224030. http://doi.org/10.1128/JB.185.14.4022-4030.2003
Weinbauer, M. G. (2004). Ecology of prokaryotic viruses. FEMS Microbiology Reviews, 28(2), 127181. http://doi.org/10.1016/j.femsre.2003.08.001
Weitz, J. S., Hartman, H., and Levin, S. A. (2005). Coevolutionary arms races between bacteria and bacteriophage. Proceedings of the National Academy of Sciences, 102(27), 95359540. http://doi.org/10.1073/pnas.0504062102
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., and Hochstrasser, D. F. (1999). Protein identification and analysis tools in the ExPASy server. Methods Molecular Biology, 112(February), 531552.
Wu, G. A., Jun, S.-R., Sims, G. E., and Kim, S.-H. (2009). Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. Proceedings of the National Academy of Sciences, 106(31), 1282612831. http://doi.org/10.1073/pnas.0905115106
Yang, K. K., Wu, Z., Bedbrook, C. N., and Arnold, F. H. (2017). Learned Protein Embeddings for Machine Learning. Bioinformatics.
Yao, G. W., Duarte, I., Le, T. T., Carmody, L., LiPuma, J. J., Young, R., and Gonzalez, C. F. (2017). A Broad-host-range Tailocin from Burkholderia cenocepacia, 83(10), 117.
Youle, M. (2017). Thinking like a phage.
Yu, Z. G., Chu, K. H., Li, C. P., Anh, V., Zhou, L. Q., and Wang, R. W. (2010). Whole- proteome phylogeny of large dsDNA viruses and parvoviruses through a composition vector method related to dynamical language model. BMC Evolutionary Biology, 10(1). http://doi.org/10.1186/1471-2148-10-192
Zhang, Q. (2016). Strategies for Identifying the Optimal Length of K- mer in a Viral Phylogenomic Analysis using Genomic Alignment-free Method.
Zhou, Q., and Liu, J. S. (2008). Extracting sequence features to predict protein - DNA interactions: A comparative study. Nucleic Acids Research, 36(12), 41374148. http://doi.org/10.1093/nar/gkn361
Zhou, S. T., Liu, R., Zhao, X., Huang, C. H., and Wei, Y. Q. (2011). Viral proteomics: The emerging cutting-edge of virus research. Science China Life Sciences, 54(6), 502512. http://doi.org/10.1007/s11427-011-4177-7
Zillig, W., Fujiki, H., Blum, W., Janekovi, D., Schweiger, M., Rahmsdorf, H., Hirsch-Kauffmann, M. (1975). In vivo and in vitro phosphorylation of DNA-dependent RNA polymerase of Escherichia coli by bacteriophage-T7-induced protein kinase. Proceed- ings of the National Academy of Sciences of the United States of America, 72(7), 250610. Retrieved https://www.ncbi.nlm.nih.gov/pubmed/1101258
Zinder, N. D. (1958). Lysogenization and superinfection immunity in Salmonella. Virology, 5(2), 291326. http://doi.org/10.1016/0042-6822(58)90025-4








