
OBSTAKELS VAN TTP: Towards better diagnosis and treatment of Thrombotic Thrombocytopenic Purpura
Niet alleen de naam is een mondvol, maar ook de ziekte zelf laat zich niet makkelijk temmen. TTP is zonder behandeling fataal in 90% van de gevallen en zelfs met behandeling blijft het sterftecijfer 20%. Daarnaast zullen 40% van de patiënten hervallen. Voor vele patiënten blijft het dus niet bij een eenmalige acute episode, maar kan de ziekte gezien worden als een chronische aandoening met terugkerende (plotse) levensbedreigende aanvallen. Het uitstellen van de behandeling kan extreme gevolgen hebben, zoals ernstige orgaanschade, en kan zelfs leiden tot overlijden van de patiënt. Daarom vergt een acute TTP-aanval onmiddellijke hospitalisering.
Wat is de Oorzaak?
VWF (von Willebrand factor) is een essentieel molecule dat circuleert in ons bloed. Het werkt als een stukje kleefband dat bloedplaatjes bindt en zich vasthecht op plaatsen in het bloedvat waar er schade is. De binding van VWF, en dus ook bloedplaatjes, zorgt ervoor dat het bloed gestelpt wordt en geen bloed wegsijpelt naar de omliggende weefsels. Je kan het zien als een korstje aan de binnenkant van het bloedvat dat ervoor zorgt dat de wonde snel geneest.
VWF wordt echter geproduceerd als een heel lang stuk kleefband, terwijl er bij wondschade slechts een klein stukje nodig is. Deze lange stukken kleefband zijn gevaarlijk omdat ze spontaan kunnen openrollen en bloedplaatjes binden. Er wordt dan een bloedklonter gevormd, ook al is het bloedvat niet beschadigd. Om dit te voorkomen, werkt het eiwit ADAMTS13 als een schaar die de lange stukken VWF in kleine stukjes kleefband knipt. ADAMTS13 zorgt er dus voor dat er geen bloedklonters worden gevormd.
TTP wordt veroorzaakt door een tekort aan ADAMTS13. De oorzaak van dit tekort kan een mutatie in het ADAMTS13-gen zijn of de vorming van antilichamen tegen ADAMTS13.
Wat zijn de symptomen?
Het gevolg van een ADAMTS13-deficiëntie is dus de vorming van kleine Trombi. Deze trombi sluiten kleine bloedvaten af van bloed en bijgevolg zuurstof. Dit zuurstoftekort kan resulteren in orgaanfalen en uiteindelijk leiden tot de dood van de patiënt. TTP-patiënten vertonen vooral symptomen gelinkt aan hersenschade, hartfalen en problemen met het darm-maagstelsel, alsook de nieren.
Daarnaast worden TTP-patiënten geconfronteerd met Trombocytopenie. Dit betekent dat ze een tekort hebben aan vrije bloedplaatjes (trombocyten) in hun bloed.
Bijgevolg vertonen sommige TTP-patiënten paarse vlekken op de huid, die Purpura worden genoemd (Figuur 1). Bloedplaatjes helpen namelijk het bloed te stelpen wanneer een bloedvat beschadigd is. Wanneer er een tekort is aan bloedplaatjes, gebeurt dit niet en ontstaat er een lekkage van bloed uit de beschadigde bloedvaten.
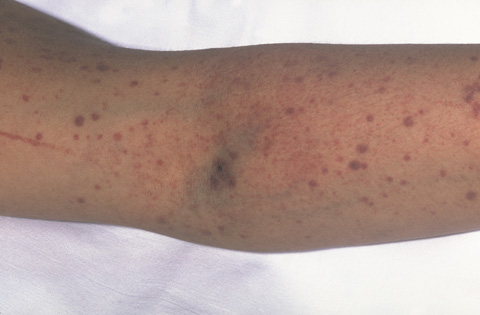
Figuur 1. Patiënt met purpura (grotere vlekken) en petechiën (rood-paarse stippen). Dit zijn kleine bloeduitstortingen in de huid. (Geraapleegd van National Heart, Lung, and Blood Institute; National Institutes of Health; U.S. Department of Health and Human Services. https://www.nhlbi.nih.gov/health-topics/thrombotic-thrombocytopenic-purpura.)
Als al dit gekend is… wat is dan het probleem?
Eerst en vooral: sommige zaken zijn nog niet gekend of niet helemaal duidelijk. In het onderzoek leidt het antwoord op de ene vraag vaak naar meerdere nieuwe vragen en dat is bij deze ziekte niet anders.
Het is belangrijk voor TTP dat er nog verder onderzoek wordt uitgevoerd naar:
- Factoren of manieren die het diagnoseproces kunnen vergemakkelijken. Momenteel is de diagnoseprocedure uiterst complex omdat de symptomen van TTP overlappen met die van andere ziektes. Bovendien kan TTP enkel met zekerheid gediagnosticeerd worden als er onderzoek wordt gedaan naar de activiteit van ADAMTS13, en het kan even wachten zijn op deze resultaten (vaak worden deze testen uitgevoerd in gespecialiseerde labo’s en niet in het ziekenhuis zelf). Kortom, deze complexiteit kan leiden tot een verkeerde diagnose en vertragingen in TTP-diagnose.
- Factoren die de ernst van een aanval kunnen verduidelijken en kunnen voorspellen wanneer een patiënt zal hervallen.
- Betere behandelingsmethodes, omdat TTP een chronische levensbedreigende ziekte is waarbij het sterftecijfer nog altijd 20% is.
Wat brengt deze studie bij?
Drie ADAMTS13-biomarkers die belangrijk zijn voor de diagnose en prognose van TTP-patiënten werden aangehaald in deze studie, namelijk:
- ADAMTS13 activiteit, wat nodig is voor het diagnosticeren van TTP. Er bestaan testen die kunnen meten “hoe actief ADAMTS13 is” en bepalen of deze functioneel is in de patiënt of niet. Echter, deze zijn duur en vragen veel vaardigheid, en worden daarom meestal niet in het ziekenhuis zelf uitgevoerd, waardoor kostbare tijd wordt verspild.
- anti-ADAMTS13 antilichamen, om een onderscheid te kunnen maken tussen patiënten waarvan de ADAMTS13-deficiëntie veroorzaakt wordt door antilichamen tegen ADAMTS13 en patiënten die een mutatie hebben in het ADAMTS13-gen. Dit onderscheid is belangrijk omdat deze patiënten verschillend behandeld worden.
- ADAMTS13 antigen, waarvan eerder onderzoek aangeeft dat dit wellicht een goede prognostische factor is en waarnaar verder onderzoek gewenst is.
In deze studie werd voor elk van deze markers een eenvoudige test ontwikkeld of verder uitgewerkt.
Ten slotte werd een klinisch relevante gentherapie voorgesteld voor patiënten met TTP, veroorzaakt door een mutatie in het ADAMTS13-gen. Bij gentherapie wordt genetisch materiaal ingebracht in de cellen, bij de voorgestelde methode is dat in de spiercellen. Dit genetisch materiaal zal in de cel gebruikt worden om een eiwit te produceren waarvan de patiënt (zonder de gentherapie) een tekort heeft, in dit geval ADAMTS13.
Om deze gentherapie te onderzoeken, werd genetisch materiaal gecreëerd, namelijk een DNA plasmide die murien (van de muis) ADAMTS13-DNA bevat. Er werd in vitro (in spiercellen die gekweekt worden in het labo) geverifieerd of dit plasmide zorgt voor de aanmaak van ADAMTS13. In de toekomst zal dit plasmide geïntroduceerd worden in spiercellen van muizen die een tekort hebben aan ADAMTS13. Zo wordt onderzocht of dit plasmide ook in vivo geproduceerd wordt en of deze gentherapie de muizen beschermt tegen TTP.
Bibliografie
Aihara, H., & Miyazaki, J. (1998). Gene transfer into muscle by electroporation in vivo. Nat. Biotechnol., 16(9), 867–870.
Alwan, F., Vendramin, C., Vanhoorelbeke, K., et al. (2017). Presenting ADAMTS13 antibody and antigen levels predict prognosis in immune-mediated thrombotic thrombocytopenic purpura. Blood, 130(4), 466–471.
Amorosi, E. L., & Ultmann, J. E. (1966). Thrombotic thrombocytopenic purpura: report of 16 cases and review of the literature. Medicine, 45(2), 139–160.
Anderson, P. J., Kokame, K., & Sadler, J. E. (2006). Zinc and calcium ions cooperatively modulate ADAMTS13 activity. J. Biol. Chem., 281(2), 850–857.
André, F., & Mir, L. M. (2004). DNA electrotransfer: its principles and an updated review of its therapeutic applications. Gene Ther., 11(Suppl 1), S33–S42.
Blombäck, M., Eikenboom, J., Lane, D., Denis, C., & Lillicrap, D. (2012). von Willebrand disease biology. Haemophilia, 18(Suppl 4), 141–147.
Blombery, P., & Scully, M. (2014). Management of thrombotic thrombocytopenic purpura : current perspectives. J. Blood Med., 5, 15–23.
Boulpaep, E. L. (2017). Blood. In Boron, W. F., & Boulpaep, E. L. (Eds.), Medical Physiology (pp. 429–446). Philadelphia, PA: Elsevier.
Cataland, S. R., Yang, S., Witkoff, L., Kraut, E. H., Lin, S., George, J. N., & Wu, H. M. (2009). Demographic and ADAMTS13 biomarker data as predictors of early recurrences of idiopathic thrombotic thrombocytopenic purpura. Eur. J. Haematol., 83(6), 559–564.
Chapin, J. C., & Hajjar, K. A. (2015). Fibrinolysis and the control of blood coagulation. Blood Reviews, 29(1), 17–24.
Coppo, P., Cuker, A., & George, J. N. (2019). Thrombotic thrombocytopenic purpura : Toward targeted therapy and precision medicine. Res. Pract. Thromb. Haemost., 3(1), 26–37.
Crawley, J. T. B., de Groot, R., Xiang, Y., Luken, B. M., & Lane, D. A. (2011). Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood, 118(12), 3212–3221.
De Cock, E., Hermans, C., De Raeymaecker, J., et al. (2015). The novel ADAMTS13-p.D187H mutation impairs ADAMTS13 activity and secretion and contributes to thrombotic thrombocytopenic purpura in mice. J. Thromb. Haemost., 13(2), 283–292.
Deforche, L., Tersteeg, C., Roose, E., et al. (2016). Generation of anti-murine ADAMTS13 antibodies and their application in a mouse model for acquired thrombotic thrombocytopenic purpura. PLoS ONE, 11(8), 1-15.
de Groot, R., Lane, D. A., & Crawley, J. T. B. (2010). The ADAMTS13 metalloprotease domain: roles of subsites in enzyme activity and specificity. Blood, 116(16), 3064–3072.
de Groot, R., Lane, D. A., & Crawley, J. T. B. (2015). The role of the ADAMTS13 cysteine-rich domain in VWF binding and proteolysis. Blood, 125(12), 1968–1975.
de Laat, B., van Berkel, M., Urbanus, R. T., Siregar, B., de Groot, P. G., Gebbink, M. F., & Maas, C. (2011). Immune responses against domain I of β2-glycoprotein I are driven by conformational changes: Domain I of β2-glycoprotein I harbors a cryptic immunogenic epitope. Arthritis Rheum., 63(12), 3960–3968.
Ercig, B., Wichapong, K., Reutelingsperger, C. P. M., Vanhoorelbeke, K., Voorberg, J., & Nicolaes, G. A. F. (2018). Insights into 3D Structure of ADAMTS13: A Stepping Stone towards Novel Therapeutic Treatment of Thrombotic Thrombocytopenic Purpura. Thromb Haemost., 118(1), 28–41.
Feng, Y., Li, X., Xiao, J., Li, W., Liu, J., Zeng, X., Chen, X., & Chen, S. (2016). ADAMTS13: more than a regulator of thrombosis. International Journal of Hematology, 104(5), 534–539.
Ferrari, S., Palavra, K., Gruber, B., et al. (2014). Persistence of circulating ADAMTS13-specific immune complexes in patients with acquired thrombotic thrombocytopenic purpura. Haematologica, 99(4), 779–787.
Feys, H. B., Liu, F., Dong, N., et al. (2006). ADAMTS-13 plasma level determination uncovers antigen absence in acquired thrombotic thrombocytopenic purpura and ethnic differences. J. Thromb. Haemost., 4(5), 955–962.
Finley, A., & Greenberg, C. (2013). Heparin sensitivity and resistance: management during cardiopulmonary bypass. Anesth. Analg., 116(6), 1210–1222.
Furlan, M., Robles, R., & Lämmle, B. (1996). Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood, 87(10), 4223–4234.
George, J. N. (2010). How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood, 116(20), 4060–4069.
Grall, M., Azoulay, E., Galicier, L., et al. (2017). Thrombotic thrombocytopenic purpura misdiagnosed as autoimmune cytopenia: Causes of diagnostic errors and consequence on outcome. Experience of the French thrombotic microangiopathies reference centre. Am. J. Hematol., 92(4), 381–387.
Grillberger, R., Casina, V. C., Turecek, P. L., Zheng, X. L., Rottensteiner, H., & Scheiflinger, F. (2014). Anti-ADAMTS13 IgG autoantibodies present in healthy individuals share linear epitopes with those in patients with thrombotic thrombocytopenic purpura. Haematologica, 99(4), e58–e60.
Hollevoet, K., De Smidt, E., Geukens, N., & Declerck, P. (2018). Prolonged in vivo expression and anti-tumor response of DNA- based anti-HER2 antibodies. Oncotarget, 9(17), 13623–13636.
Hollevoet, K., & Declerck, P. J. (2017). State of play and clinical prospects of antibody gene transfer. Journal of Translational Medicine, 15(131).
James, P. D., & Lillicrap, D. (2013). The molecular characterization of von Willebrand disease: good in parts. Br. J. Haematol., 161(2), 166–176.
Jin, S., Xiao, J., Bao, J., Zhou, S., Wright, J. F., & Zheng, X. L. (2013). AAV-mediated expression of an ADAMTS13 variant prevents shigatoxin-induced thrombotic thrombocytopenic purpura. Blood, 121(19), 3825–3829.
Joly, B. S., Coppo, P., & Veyradier, A. (2017). Thrombotic thrombocytopenic purpura. Blood, 129(21), 2836–2847. (a)
Joly, B. S., Vanhoorelbeke, K., & Veyradier, A. (2017). Understanding therapeutic targets in thrombotic thrombocytopenic purpura. Intensive Care Med., 43(9), 1398–1400. (b)
Kato, S., Hiura, H., Fujimura, Y., & Matsumoto, M. (2012). European Patent No. EP1852442B1. Retrieved from https://patents.google.com/patent/EP1852442B1/en.
Kato, S., Matsumoto, M., Matsuyama, T., Isonishi, A., Hiura, H., & Fujimura, Y. (2006). Novel monoclonal antibody-based enzyme immunoassay for determining plasma levels of ADAMTS13 activity. Transfusion. 46(8), 1444–1452.
Katsumi, A., Tuley, E. A., Bodó, I., & Sadler, J. E. (2000). Localization of Disulfide Bonds in the Cystine Knot Domain of Human von Willebrand Factor. J. Biol. Chem., 275(33), 25585–25594.
Knöbl, P. (2018). Thrombotic thrombocytopenic purpura. Memo, 11(3), 220–226.
Kokame, K., Nobe, Y., Kokubo, Y., Okayama, A., & Miyata, T. (2005). FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br. J. Haematol., 129(1), 93–100.
Koyama, N., Matsumoto, M., Tamaki, S., Yoshikawa, M., Fujimura, Y., & Kimura, H. (2012). Reduced larger von Willebrand factor multimers at dawn in OSA plasmas reflect severity of apnoeic episodes. Eur. Respir. J., 40(3), 657–664.
Kreimann, M., Brandt, S., Krauel, K., Block, S., Helm, C. A., Weitschies, W., Greinacher, A., & Delcea, M. (2014). Binding of anti-platelet factor 4/heparin antibodies depends on the thermodynamics of conformational changes in platelet factor 4. Blood, 124(15), 2442–2449.
Kremer Hovinga, J. A., Vesely, S. K., Terrell, D. R., Lämmle, B., & George, J. N. (2010). Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood, 115(8), 1500–1511.
Kremer Hovinga, J. A., & Voorberg, J. (2012). Improving on nature: redesigning ADAMTS13. Blood, 119(16), 3654–3655.
Kremer Hovinga, J. A., & Lämmle, B. (2012). Role of ADAMTS13 in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. Hematol. Am. Soc. Hematol. Educ. Progr. 2012, 1, 610–616.
Kremer Hovinga, J. A., Coppo, P., Lämmle, B., Moake, J. L., Miyata, T., & Vanhoorelbeke, K. (2017). Thrombotic thrombocytopenic purpura. Nat. Rev. - Dis. Prim., 3, 1-17.
Laje, P., Shang, D., Cao, W., et al. (2009). Correction of murine ADAMTS13 deficiency by hematopoietic progenitor cell-mediated gene therapy. Blood, 113(10), 2172–2180.
Lam, J. K., Chion, C. K. N. K., Zanardelli, S., Lane, D. A., & Crawley, J. T. B. (2007). Further characterization of ADAMTS-13 inactivation by thrombin. J. Thromb. Haemost., 5(5), 1010–1018.
Lämmle, B., Kremer Hovinga, J. A., & Alberio, L. (2005). Thrombotic thrombocytopenic purpura. J. Thromb. Haemost., 3(8), 1663–1675.
Lenting, P. J., Christophe, O. D., & Denis, C. V. (2015). von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood, 125(13), 2019–2028.
Levy, G. G., Nichols, W. C., Lian, E. C., et al. (2001). Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature, 413(6855), 488–494.
Lillicrap, D. (2013). von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood, 122(23), 3735–3740.
Liu-Chen, S., Connolly, B., Cheng, L., Subramanian, R. R., & Han, Z. (2018). mRNA treatment produces sustained expression of enzymatically active human ADAMTS13 in mice. Sci. Rep., 8, 7859.
Lopes da Silva, M., & Cutler, D. F. (2016). von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cells. Blood, 128(2), 277–285.
Lotta, L. A., Garagiola, I., Palla, R., Cairo, A., & Peyvandi, F. (2010). ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum. Mutat., 31(1), 11–19.
Luo, G., Ni, B., Yang, X., & Wu, Y. (2012). von Willebrand Factor: More Than a Regulator of Hemostasis and Thrombosis. Acta Haematol., 128(3), 158–169.
Mancini, I., Ferrari, B., Valsecchi, C., Pontiggia, S., Fornili, M., Biganzoli, E., & Peyvandi, F. (2017). ADAMTS13-specific circulating immune complexes as potential predictors of relapse in patients with acquired thrombotic thrombocytopenic purpura. Eur. J. Intern. Med., 39, 79–83.
Martino, S., Jamme, M., Deligny, C., et al. (2016). Thrombotic Thrombocytopenic Purpura in Black People: Impact of Ethnicity on Survival and Genetic Risk Factors. PLOS ONE, 11(7), 3-12.
Moake, J. L., Rudy, C. K., Troll, J. H., Weinstein, M. J., Colannino, N. M., Azocar, J., Seder, R. H., Suchen L. H., & Deykin, D. (1982). Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N. Engl. J. Med., 307(23), 1432–1435.
Mombaerts, P., Iacomini, J., Johnson, R. S., Herrup, K., Tonegawa, S., & Papaioannouo, V. E. (1992). RAG-1-Deficient Mice Have No Mature B and T Lymphocytes. Cell, 68, 869–877.
Monroe, D. M., & Hoffman, M. (2006). What Does It Take to Make the Perfect Clot? Arterioscler. Thromb. Vasc. Biol., 26(1), 41–48.
Motto, D. G., Chauhan, A. K., Zhu, G., et al. (2005). Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J. Clin. Invest., 115(10), 2752–2761.
Muia, J., Gao, W., Haberichter, S. L., Dolatshahi, L., Zhu, J., Westfield, L. A., Covill, S. C., & Sadler, J. E. (2013). An optimized fluorogenic ADAMTS13 assay with increased sensitivity for the investigation of patients with thrombotic thrombocytopenic purpura. J. Thromb. Haemost., 11(8), 1511–1518.
Muia, Joshua, Zhu, J., Gupta, G., et al. (2014). Allosteric activation of ADAMTS13 by von Willebrand factor. Proc. Natl. Acad. Sci., 111(52), 18584–18589.
Müller, J. P., Mielke, S., Löf, A., et al. (2016). Force sensing by the vascular protein von Willebrand factor is tuned by a strong intermonomer interaction. Proc. Natl. Acad. Sci., 113(5), 1208–1213.
Murphy, K., & Weaver, C. (2017). Janeway’s immunobiology (9th ed.). New York, NY: Garland Science.
Niiya, M., Endo, M., Shang, D., et al. (2009). Correction of ADAMTS13 Deficiency by In Utero Gene Transfer of Lentiviral Vector encoding ADAMTS13 Genes. Mol. Ther., 17(1), 34–41.
Peyvandi, F., Scully, M., Kremer Hovinga, J. A., et al. (2016). Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med., 374(6), 511–522.
Plautz, W. E., Raval, J. S., Dyer, M. R., Rollins-Raval, M. A., Zuckerbraun, B. S., & Neal, M. D. (2018). ADAMTS13: origins, applications, and prospects. Transfusion, 58(10), 2453-2462.
Rauch, A., Wohner, N., Christophe, O. D., Denis, C. V., Susen, S., & Lenting, P. J. (2013). On the versatility of von Willebrand Factor. Mediterr J Hematol Infect Dis., 5(1), e2013046.
Raval, J. S., Padmanabhan, A., Kremer Hovinga, J. A., & Kiss, J. E. (2015). Development of a clinically significant ADAMTS13 inhibitor in a patient with hereditary thrombotic thrombocytopenic purpura. Am. J. Hematol., 90(1), E22.
Reininger, A. J. (2008). Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia, 14(Suppl 5), 11–26.
Rieger, M., Ferrari, S., Kremer Hovinga, J. A., et al. (2006). Relation between ADAMTS13 activity and ADAMTS13 antigen levels in healthy donors and patients with thrombotic microangiopathies (TMA). Thromb Haemost., 95(2), 212–220.
Roose, E., Schelpe, A., Joly, B. S., et al. (2018). An open conformation of ADAMTS-13 is a hallmark of acute acquired thrombotic thrombocytopenic purpura. J. Thromb. Haemost., 16(2), 378–388. (a)
Roose, E., Vidarsson, G., Kangro, K., et al. (2018). Anti-ADAMTS13 Autoantibodies against Cryptic Epitopes in Immune-Mediated Thrombotic Thrombocytopenic Purpura. Thromb Haemost., 118(10), 1729–1742. (b)
Ruggeri, Z. M. (2002). Platelets in atherothrombosis. Mayo Clinic Proceedings. Mayo Clinic, 8(11), 1227–1234.
Sadler, J. E. (2002). A new name in thrombosis, ADAMTS13. Proc. Natl. Acad. Sci., 99(18), 11552–11554.
Sadler, J. E. (2015). What’s new in the diagnosis and pathophysiology of thrombotic thrombocytopenic purpura. Hematol. Am. Soc. Hematol. Educ. Progr. 2015, 1, 631–636.
Sadler, J. E. (2017). Pathophysiology of thrombotic thrombocytopenic purpura. Blood, 130(10), 1181–1188.
Sadler, J. E., Muia, J., & Weiqiang, G. (2013). United States Patent No. US20130023004A1. Retrieved from https://patents.google.com/patent/US20130023004A1/en.
Sadler, J. E., Muia, J., & Weiqiang, G. (2014). United States Patent No. US8663912B2. Retrieved from https://patents.google.com/patent/US8663912B2/en.
Sanders, Y. V, Groeneveld, D., Meijer, K., et al. (2015). von Willebrand factor propeptide and the phenotypic classification of von Willebrand disease. Blood, 125(19), 3006–3013.
Schelpe, A., Orlando, C., Ercig, B., et al. (2018). Child-onset thrombotic thrombocytopenic purpura caused by p.R498C and p.G259PfsX133 mutations in ADAMTS13. Eur. J. Haematol., 101(2), 191–199.
Schelpe, A. (2018). Anti-ADAMTS13 antibodies: Insights into ADAMTS13 biology and thrombotic thrombocytopenic purpura pathophysiology using novel anti-ADAMTS13 antibodies (Doctoral dissertation). Leuven, Belgium: KU Leuven, Faculty of Science.
Schiviz, A., Wuersch, K., Piskernik, C., Dietrich, B., Hoellriegl, W., Rottensteiner, H., Scheiflinger, F., Schwarz, H.P., & Muchitsch, E. M. (2012). A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood, 119(25), 6128–6135.
South, K., Luken, B. M., Crawley, J. T. B., Phillips, R., Thomas, M., Collins, R. F., Deforche, L., Vanhoorelbeke, K., & Lane, D. A. (2014). Conformational activation of ADAMTS13. Proc. Natl. Acad. Sci., 111(52), 18578–18583.
South, K., Freitas, M. O., & Lane, D. A. (2017). A model for the conformational activation of the structurally quiescent metalloprotease ADAMTS13 by von willebrand factor. J. Biol. Chem., 292(14), 5760–5769.
Stockschlaeder, M., Schneppenheim, R., & Budde, U. (2014). Update on von Willebrand factor multimers: Focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul. Fibrinolysis, 25(3), 206–216.
Tersteeg, C., Schiviz, A., De Meyer, S. F., Plaimauer, B., Scheiflinger, F., Rottensteiner, H., & Vanhoorelbeke, K. (2015). Potential for Recombinant ADAMTS13 as an Effective Therapy for Acquired Thrombotic Thrombocytopenic Purpura. Arterioscler. Thromb. Vasc. Biol., 35(11), 2336–2342.
Thomas, M. R., de Groot, R., Scully, M. A., & Crawley, J. T. B. (2015). Pathogenicity of Anti-ADAMTS13 Autoantibodies in Acquired Thrombotic Thrombocytopenic Purpura. EBioMedicine, 2(8), 942–952.
Trionfini, P., Tomasoni, S., Galbusera, M., Motto, D., Longaretti, L., Corna, D., Remuzzi, G., & Benigni, A. (2009). Adenoviral-mediated gene transfer restores plasma ADAMTS13 antigen and activity in ADAMTS13 knockout mice. Gene Ther., 16(11), 1373–1379.
Truett, G. E., Heeger, P., Mynatt, R. L., Truett, A. A., Walker, J. A., & Warman, M. L. (2000). Preparation of PCR-Quality Mouse Genomic DNA with Hot Sodium Hydroxide and Tris (HotSHOT). BioTechniques, 29(1), 52–54.
Tsai, H. (1996). Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood, 87(10), 4235–4244.
Tsai, H. (2012). von Willebrand Factor, Shear Stress, and ADAMTS13 in Hemostasis and Thrombosis. ASAIO J., 58(2), 163-169.
Underwood, M., Peyvandi, F., Garagiola, I., Machin, S., & Mackie, I. (2016). Degradation of two novel congenital TTP ADAMTS13 mutants by the cell proteasome prevents ADAMTS13 secretion. Thrombosis Research, 147, 16–23.
Vanhoorelbeke, K., Cauwenberghs, N., Vandecasteele, G., Vauterin, S., & Deckmyn, H. (2002). A Reliable von Willebrand Factor: Ristocetin Cofactor Enzyme-Linked Immunosorbent Assay to Differentiate between Type 1 and Type 2 von Willebrand Disease. Seminars in Thrombosis and Hemostasis, 28(2), 161–166.
Vanhoorelbeke, K., & De Meyer, S. F. (2013). Animal models for thrombotic thrombocytopenic purpura. J Thromb Haemost, 11(Suppl 1), 2–10.
Verhenne, S., Vandeputte, N., Pareyn, I., Izsvák, Z., Rottensteiner, H., Deckmyn, H., De Meyer, S. F., & Vanhoorelbeke, K. (2017). Long-term prevention of congenital thrombotic thrombocytopenic purpura in ADAMTS13 knockout mice by sleeping beauty transposon-mediated gene therapy. Arterioscler. Thromb. Vasc. Biol., 37(5), 836–844.
Versteeg, H. H., Heemskerk, J. W. M., Levi, M., & Reitsma, P. H. (2013). New Fundamentals in Hemostasis. Physiol. Rev., 93(1), 327–358.
Yang, S., Jin, M., Lin, S., Cataland, S., & Wu, H. (2011). ADAMTS13 activity and antigen during therapy and follow-up of patients with idiopathic thrombotic thrombocytopenic purpura: correlation with clinical outcome. Haematologica, 96(10), 1521–1527.
Zander, C. B., Cao, W., & Zheng, X. L. (2015). ADAMTS13 and von Willebrand factor interactions. Curr. Opin. Hematol., 22(5), 452–459.
Zheng, X., Chung, D., Takayama, T. K., Majerus, E. M., Sadler, J. E., & Fujikawa, K. (2001). Structure of von Willebrand Factor-cleaving Protease (ADAMTS13), a Metalloprotease Involved in Thrombotic Thrombocytopenic Purpura. J. Biol. Chem., 276(44), 41059–41063.
Zheng, X. L. (2013). Structure-function and regulation of ADAMTS-13 protease. J. Thromb. Haemost., 11, 11–23.
Zheng, X. L. (2015). ADAMTS13 and von Willebrand Factor in Thrombotic Thrombocytopenic Purpura. Annu. Rev. Med., 66(1), 211–225.
Zhou, Y., Eng, E. T., Zhu, J., Lu, C., Walz, T., & Springer, T. A. (2012). Sequence and structure relationships within von Willebrand factor. Blood, 120(2), 449–458.






