
Bacteriën als sluitstuk van onze stikstofkringloop?
2 500 000 TJ, ofwel 13 keer de jaarlijkse capaciteit van het Belgisch kernenergiepark. Zo veel energie wordt er elk jaar verbruikt door de ammoniakindustrie. Nochtans is ammoniak een onmisbare grondstof voor de kunstmest die u in uw dagelijkse portie voedsel voorziet. Kan dit duurzamer?
Stikstoffixatie: de stap te veel?
Ammoniak wordt vandaag geproduceerd via ‘stikstoffixatie’, ofwel het vastzetten van stikstofgas in stoffen die gewassen gemakkelijker kunnen opnemen. Het huidige productieproces gebruikt hiervoor waterstofgas verkregen uit aardgas en stikstofgas uit lucht. Daardoor neemt het wel 5 % van de globale aardgasvraag en 1 % van de totale CO2-emissies voor zijn rekening. Deze gefixeerde stikstof belandt uiteindelijk in ons afval. Nu, u zou kunnen zeggen dat dit een noodzakelijk kwaad is, maar tijdens de afvalverwerking wordt een deel van deze stikstof terug in stikstofgas ‘gedefixeerd’. Zonde van de energie die verbruikt is tijdens de productie.
Wat als we gefixeerde stikstof konden recycleren en zo het fixatieproces overslaan? Mijn thesis bouwt verder op een idee dat bacteriën inschakelt om afvalstikstof terug om te zetten in ammoniak en zo de menselijke stikstofcyclus rond te maken. De stikstofcomponenten in ons afval zijn vooral proteïnes. Bacteriën zoals de Bacillus subtilis kunnen deze grote molecules afbreken tot hun afzonderlijke bouwstenen, de aminozuren. De microbe hergebruikt deze dan om nieuwe proteïnes en cellen aan te maken of breekt ze volledig af voor zijn eigen energievoorziening, wat CO2 en ammoniak oplevert.
Vechten tegen Darwin
Jammer genoeg is er een stevig obstakel. De evolutietheorie van Darwin, u hoeft daar waarschijnlijk geen inleiding bij. Een micro-organisme zoals de Bacillus subtilis tracht de soort te laten overleven o.a. door zo veel mogelijk cellen aan te maken, want dan zijn de kansen groter dat er minstens één het overleeft. Alle twintig types aminozuren zijn daarvoor nodig, maar vaak moet het ene type deels in het andere worden omgezet, wat dikwijls ammoniak verbruikt. Het eventuele overschotje aan ammoniak is voor ons.
Het metabolisme is dus cruciaal. Dat is het ensemble aan chemische reacties dat een organisme uitvoert. Om ammoniak over te houden, moet het verbruik voor de opbouw van cellen beperkt worden en/of de ammoniakproducerende reacties versterkt. Elke reactie heeft zijn eigen biologische katalysator, het enzym, waarvan de bouwplannen in het bijhorende gen in het DNA vervat zijn. De reactiesnelheid kan gecontroleerd worden o.a. door de expressie van dat gen te regelen. De centrale vraag in deze thesis is daarom in welke reacties - en bijgevolg genen – er moet ingegrepen worden om zo veel mogelijk ammoniak te verkrijgen.
Metabolische netwerkmodellen: de computer als loodgieter
Ik vond een mogelijke oplossing in het simuleren van het metabolisme via een zogenaamd ‘metabolisch netwerkmodel’. Door opeenvolgende reacties aan elkaar te linken in een diagram, is het mogelijk om het volledige metabolisme voor te stellen als één groot netwerk waarvan elke lijn afzonderlijk een reactie met zijn enzym voorstelt. Het aantal lijnen kan gemakkelijk oplopen tot boven de duizend. Vergelijk het met een groot waterleidingnetwerk waarbij elke waterstroom een reactie voorstelt en waar er op elke afzonderlijke buis een kraantje zit om de stroomsnelheid te regelen. Het is dus niet verwonderlijk dat alle reactiesnelheden in dit netwerk opmeten een lastig karwei is.
U voelt de bui al hangen: het probleem is tweeledig, zoals Figuur 1 illustreert. Eerst moet er een manier gevonden worden om alle reactiesnelheden in het netwerk te bepalen. Pas daarna kan het netwerk zelf onder handen genomen worden. Gelukkig kan de wiskunde hulp bieden bij het eerste probleem door een beredeneerde gok te doen. Het is mogelijk met een computer er een optimalisatiealgoritme op los te laten. Daarbij definieer je enerzijds het ‘objectief’, dat wat geoptimaliseerd wordt, en anderzijds beperkingen om de zoekruimte af te bakenen. Uit een vorige paragraaf weet u dat een microbe zo veel mogelijk cellen tracht aan te maken. Klinkt dat niet als een objectief? Verder zijn er nog allerlei beperkingen zoals de snelheid waarmee de bacterie aminozuren kan verwerken, de hoeveelheid aanwezige enzymen... Samenvattend, dit optimalisatiealgoritme schat welk metabolisme het micro-organisme nastreeft met de beschikbare middelen in zijn overlevingsstrijd. Het plaatst zo als het ware een wijzerplaatje op elke leiding van het waternetwerk.
Nu kunnen we doelgericht aan het metabolisme zelf sleutelen. De vraag hierbij is aan welke figuurlijke kraantjes te draaien opdat de ammoniakstroom zo groot mogelijk wordt. Via de methode hierboven wordt telkens het effect van een ingreep bepaald. Twee nieuwe algoritmes testen zo verschillende combinaties uit, elk volgens hun eigen strategie. Het eerste zoekt met brute rekenkracht één ingreep die de grootste verbetering teweeg brengt, zet deze vast en herhaalt dit voor elke nieuwe versie van het netwerk. Het andere simuleert biologische evolutie, waarbij een hogere ammoniakproductie als superieur wordt gezien. Door een samenspel van mutaties en paringsvoorkeuren onthult het de beste genetische veranderingen.
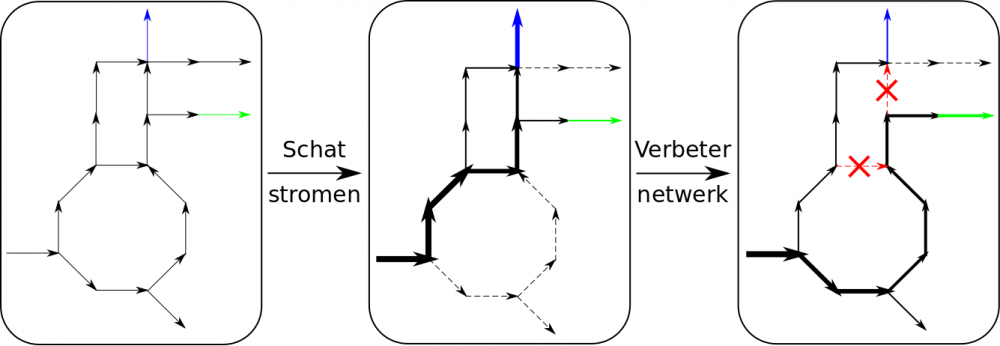
Figuur 1: Fictief netwerk met illustratie van de optimalisatiemethode. De cel optimaliseert spontaan de celaanmaak (blauw), maar door ingrepen wordt de cel gedwongen daarbij zo veel mogelijk gewenst product (groen) te maken. De pijldikte is evenredig met de stroomsnelheid.
Goed begonnen...
Ondanks hun verschillende aanpak wezen beide algoritmes in dezelfde richting. Een combinatie van twee types ingrepen kan de ammoniakproductie per cel tot wel 16 % doen stijgen t.o.v. het ongewijzigde metabolisme. Het ene type produceert extra ammoniak door meer stikstofrijke aminozuren af te breken. Het andere verlaagt het ammoniakverbruik door de aanmaak van nieuwe cellen te beperken.
Als aanvullend onderzoek werd getest of het netwerkmodel experimentele productiesnelheden kan voorspellen. Het bleek dat enkele nieuwe reacties aan het netwerk dienden toegevoegd te worden omdat er stoffen werden geproduceerd die niet in het model aanwezig waren. Verdere verfijning blijft ook nodig. Dat betekent dat we nog enigszins een slag om de arm moeten houden met de optimalisatieresultaten, maar ze wijzen toch al in een duidelijke richting.
De menselijke stikstofcyclus sluiten m.b.v. microben zal zeker nog veel voeten in de aarde hebben, maar de vooruitgang van de biotechnologie houdt ook hier alvast potentieel in. Misschien kan het zo zelfs bijdragen aan een nieuwe generatie afvalverwerking en ons energie besparen.
Bibliografie
[1] Y. Mikami, H. Yoneda, Y. Tatsukami, W. Aoki, and M. Ueda, “Ammonia production from amino acid-based biomass-like sources by engineered Escherichia coli,” AMB Express, vol. 7, no. 1, 2017.
[2] M. Appl, “Ammonia,” in Ullmann’s Encyclopedia of Industrial Chemistry, pp. 904–980, Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, dec 2006.
[3] T. M. Paschkewitz, Ammonia Production at Ambient Temperature and Pressure. PhD thesis, University of Iowa, jul 2012.
[4] N. Gil-Lalaguna, Z. Afailal, M. Aznar, and I. Fonts, “Exploring the sustainable production of ammonia by recycling N and H in biological residues: Evolution of fuel-N during glutamic acid gasification,” Journal of Cleaner Production, p. 124417, oct 2020.
[5] D. G. Wernick and J. C. Liao, “Protein-based biorefining: metabolic engineering for production of chemicals and fuel with regeneration of nitrogen fertilizers,” Applied Microbiology and Biotechnology, vol. 97, pp. 1397–1406, feb 2013.
[6] Y. X. Huo, K. M. Cho, J. G. Rivera, E. Monte, C. R. Shen, Y. Yan, and J. C. Liao, “Conversion of proteins into biofuels by engineering nitrogen flux,” Nature Biotechnology, vol. 29, no. 4, pp. 346–351, 2011.
[7] K. Y. Choi, D. G. Wernick, C. A. Tat, and J. C. Liao, “Consolidated conversion of protein waste into biofuels and ammonia using Bacillus subtilis,” Metabolic Engineering, vol. 23, pp. 53–61, 2014.
[8] I. Pikaar, S. Matassa, K. Rabaey, B. L. Bodirsky, A. Popp, M. Herrero, and W. Verstraete, “Microbes and the Next Nitrogen Revolution,” Environmental Science and Technology, vol. 51, no. 13, pp. 7297–7303, 2017.
[9] Y. Tatemichi, K. Kuroda, T. Nakahara, and M. Ueda, “Efficient ammonia production from food by-products by engineered Escherichia coli,” AMB Express, vol. 10, no. 1, 2020.
[10] Y. K. Oh, B. O. Palsson, S. M. Park, C. H. Schilling, and R. Mahadevan, “Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data,” Journal of Biological
Chemistry, vol. 282, no. 39, pp. 28791–28799, 2007.
[11] C. D. Demirhan, W. W. Tso, J. B. Powell, and E. N. Pistikopoulos, “Sustainable ammonia production through process synthesis and global optimization,” AIChE Journal, vol. 65, no. 7, 2019.
[12] M. Reese, C. Marquart, M. Malmali, K. Wagner, E. Buchanan, A. McCormick, and E. L. Cussler, “Performance of a Small-Scale Haber Process,” Industrial and Engineering Chemistry Research, vol. 55, no. 13, pp. 3742–3750, 2016.
[13] S. Giddey, S. P. Badwal, and A. Kulkarni, “Review of electrochemical ammonia production technologies and materials,” International Journal of Hydrogen Energy, vol. 38, no. 34, pp. 14576–14594, 2013.
[14] T. D. Rapson, C. M. Gregg, R. S. Allen, H. Ju, C. M. Doherty, X. Mulet, S. Giddey, and C. C. Wood, “Insights into Nitrogenase Bioelectrocatalysis for Green Ammonia Production,” ChemSusChem, vol. 13, pp. 4856–4865, sep 2020.
[15] Z. Jamaludin, S. Rollings-Scattergood, K. Lutes, and C. Vaneeckhaute, “Evaluation of sustainable scrubbing agents for ammonia recovery from anaerobic digestate,” Bioresource Technology, vol. 270, no. August, pp. 596–602, 2018.
[16] S. Bastian and F. H. Arnold, Microbial Metabolic Engineering, vol. 834 of Methods in Molecular Biology. New York, NY: Springer New York, 2012.
[17] I. Massaiu, L. Pasotti, N. Sonnenschein, E. Rama, M. Cavaletti, P. Magni, C. Calvio, and M. J. Herrgård, “Integration of enzymatic data in Bacillus subtilis genome-scale metabolic model improves phenotype predictions and enables in silico design of poly-γ-glutamic acid production strains,” Microbial Cell Factories, vol. 18, no. 1, pp. 1–20, 2019.
[18] B. Ø. Palsson and A. Varma, “Metabolic Flux Balancing: Basic Concepts, Scientific and Practical Use,” Biotechnology, vol. 12, no. October, pp. 994–998, 1994.
[19] M. R. Antoniewicz, “Methods and advances in metabolic flux analysis: a mini-review,” Journal of Industrial Microbiology and Biotechnology, vol. 42, no. 3, pp. 317–325, 2015.
[20] W. Wiechert, “13C Metabolic Flux Analysis,” Metabolic Engineering, vol. 3, pp. 195–206, jul 2001.
[21] J. D. Orth, I. Thiele, and B. O. Palsson, “What is flux balance analysis?,” Nature Biotechnology, vol. 28, no. 3, pp. 245–248, 2010.
[22] G. Dantzig, Linear Programming and Extensions, vol. 48. Princeton University Press, 2016.
[23] R. Mahadevan and C. H. Schilling, “The effects of alternate optimal solutions in constraint-based genome-scale metabolic models,” Metabolic Engineering, vol. 5, no. 4, pp. 264–276, 2003.
[24] N. E. Lewis, K. K. Hixson, T. M. Conrad, J. A. Lerman, P. Charusanti, A. D. Polpitiya, J. N. Adkins, G. Schramm, S. O. Purvine, D. Lopez-Ferrer, K. K. Weitz, R. Eils, R. König, R. D. Smith, and B. Palsson, “Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models,”
Molecular Systems Biology, vol. 6, no. 390, 2010.
[25] M. W. Covert, C. H. Schilling, and B. Palsson, “Regulation of gene expression in flux balance models of metabolism,” Journal of Theoretical Biology, vol. 213, no. 1, pp. 73–88, 2001.
[26] G. Karlebach and R. Shamir, “Modelling and analysis of gene regulatory networks,” Nature Reviews Molecular Cell Biology, vol. 9, no. 10, pp. 770–780, 2008.
[27] R. Mahadevan, J. S. Edwards, and F. J. Doyle, “Dynamic Flux Balance Analysis of diauxic growth in Escherichia coli,” Biophysical Journal, vol. 83, no. 3, pp. 1331–1340, 2002.
[28] A. Varma and B. O. Palsson, “Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110,” Applied and Environmental Microbiology, vol. 60, no. 10, pp. 3724–3731, 1994.
[29] K. Höffner, S. M. Harwood, and P. I. Barton, “A reliable simulator for dynamic flux balance analysis,” Biotechnology and Bioengineering, vol. 110, no. 3, pp. 792–802, 2013.
[30] J. A. Gomez, K. Höffner, and P. I. Barton, “DFBAlab: A fast and reliable MATLAB code for dynamic flux balance analysis,” BMC Bioinformatics, vol. 15, no. 1, pp. 1–10, 2014.
[31] S. Vijayakumar, M. Conway, P. Lio, and C. Angione, “Seeing the wood for the trees: A forest of methods for optimization and omic-network integration in metabolic modelling,” Briefings in Bioinformatics, vol. 19, no. 6, pp. 1218–1235, 2017.
[32] M. J. McAnulty, J. Y. Yen, B. G. Freedman, and R. S. Senger, “Genome-scale modeling using flux ratio constraints to enable metabolic engineering of clostridial metabolism in silico,” BMC Systems Biology, vol. 6, no. 1, p. 42, 2012.
[33] A. Goelzer, J. Muntel, V. Chubukov, M. Jules, E. Prestel, R. Nölker, M. Mariadassou, S. Aymerich, M. Hecker, P. Noirot, D. Becher, and V. Fromion, “Quantitative prediction of genome-wide resource allocation in bacteria,” Metabolic Engineering, vol. 32, pp. 232–243, 2015.
[34] S. Waldherr, D. A. Oyarzún, and A. Bockmayr, “Dynamic optimization of metabolic networks coupled with gene expression,” Journal of Theoretical Biology, vol. 365, pp. 469–485, 2015.
[35] E. Gonçalves, J. Bucher, A. Ryll, J. Niklas, K. Mauch, S. Klamt, M. Rocha, and J. Saez-Rodriguez, “Bridging the layers: Towards integration of signal transduction, regulation and metabolism into mathematical models,” Molecular BioSystems, vol. 9, no. 7, pp. 1576–1583, 2013.
[36] B. J. Sánchez, C. Zhang, A. Nilsson, P. Lahtvee, E. J. Kerkhoven, and J. Nielsen, “Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints,” Molecular Systems Biology, vol. 13,
no. 8, p. 935, 2017.
[37] J. Muntel, V. Fromion, A. Goelzer, S. Maa, U. Mäder, K. Büttner, M. Hecker, and D. Becher, “Comprehensive absolute quantification of the cytosolic proteome of bacillus subtilis by data independent, parallel fragmentation in liquid chromatography/mass spectrometry (LC/MSE),” Molecular and Cellular Proteomics, vol. 13, no. 4, pp. 1008–1019, 2014.
[38] D. Davidi, E. Noor, W. Liebermeister, A. Bar-Even, A. Flamholz, K. Tummler, U. Barenholz, M. Goldenfeld, T. Shlomi, and R. Milo, “Global characterization of in vivo enzyme catalytic rates and their correspondence to in vitro kcat measurements,” Proceedings of the National Academy of Sciences of the United
States of America, vol. 113, no. 12, pp. 3401–3406, 2016.
[39] L. Jeske, S. Placzek, I. Schomburg, A. Chang, and D. Schomburg, “BRENDA in 2019: A European ELIXIR core data resource,” Nucleic Acids Research, vol. 47, no. D1, pp. D542–D549, 2019.
[40] U. Wittig, R. Kania, M. Golebiewski, M. Rey, L. Shi, L. Jong, E. Algaa, A. Weidemann, H. Sauer-Danzwith, S. Mir, O. Krebs, M. Bittkowski, E. Wetsch, I. Rojas, and W. Müller, “SABIO-RK - Database for biochemical reaction kinetics,” Nucleic Acids Research, vol. 40, no. D1, pp. 790–796, 2012.
[41] B. Zhu and J. Stülke, “SubtiWiki in 2018: From genes and proteins to functional network annotation of the model organism Bacillus subtilis,” Nucleic Acids Research, vol. 46, no. D1, pp. D743–D748, 2018.
[42] A. Nilsson, J. Nielsen, and B. O. Palsson, “Metabolic Models of Protein Allocation Call for the Kinetome,” Cell Systems, vol. 5, no. 6, pp. 538–541, 2017.
[43] H. S. Choi, S. Y. Lee, T. Y. Kim, and H. M. Woo, “In silico identification of gene amplification targets for improvement of lycopene production,” Applied and Environmental Microbiology, vol. 76, no. 10, pp. 3097–3105, 2010.
[44] C. Bro, B. Regenberg, J. Förster, and J. Nielsen, “In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production,” Metabolic Engineering, vol. 8, no. 2, pp. 102–111, 2006.
[45] C. Wang, J. Liu, H. Liu, J. Wang, and J. Wen, “A genome-scale dynamic flux balance analysis model of Streptomyces tsukubaensis NRRL18488 to predictthe targets for increasing FK506 production,” Biochemical Engineering Journal, vol. 123, pp. 45–56, 2017.
[46] D. Segrè, D. Vitkup, and G. M. Church, “Analysis of optimality in natural and perturbed metabolic networks,” Proceedings of the National Academy of Sciences of the United States of America, vol. 99, no. 23, pp. 15112–15117, 2002.
[47] T. Shlomi, O. Berkman, and E. Ruppin, “Regulatory on/off minimization of metabolic flux changes after genetic perturbations,” Proceedings of the National Academy of Sciences, vol. 102, pp. 7695–7700, may 2005.
[48] J. Kim and J. L. Reed, “RELATCH: relative optimality in metabolic networks explains robust metabolic and regulatory responses to perturbations.,” Genome biology, vol. 13, no. 9, 2012.
[49] M. L. Mo, B. Ø. Palsson, and M. J. Herrgård, “Connecting extracellular metabolomic measurements to intracellular flux states in yeast,” BMC Systems Biology, vol. 3, no. 1, p. 37, 2009.
[50] M. R. Long, W. K. Ong, and J. L. Reed, “Computational methods in metabolic engineering for strain design,” Current Opinion in Biotechnology, vol. 34, pp. 135–141, 2015.
[51] H. Alper, Y. S. Jin, J. F. Moxley, and G. Stephanopoulos, “Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli,” Metabolic Engineering, vol. 7, no. 3, pp. 155–164, 2005.
[52] A. P. Burgard, P. Pharkya, and C. D. Maranas, “OptKnock: A Bilevel Programming Framework for Identifying Gene Knockout Strategies for Microbial Strain Optimization,” Biotechnology and Bioengineering, vol. 84, no. 6, pp. 647–657, 2003.
[53] J. Kim, J. L. Reed, and C. T. Maravelias, “Large-Scale Bi-Level strain design approaches and Mixed-Integer programming solution techniques,” PLoS ONE, vol. 6, no. 9, 2011.
[54] S. Ren, B. Zeng, and X. Qian, “Adaptive bi-level programming for optimal gene knockouts for targeted overproduction under phenotypic constraints,” BMC Bioinformatics, vol. 14, no. Suppl 2, 2013.
[55] N. Tepper and T. Shlomi, “Predicting metabolic engineering knockout strategies for chemical production: Accounting for competing pathways,” Bioinformatics, vol. 26, no. 4, pp. 536–543, 2009.
[56] K. R. Patil, I. Rocha, J. Förster, and J. Nielsen, “Evolutionary programming as a platform for in silico metabolic engineering,” BMC Bioinformatics, vol. 6, pp. 1–12, 2005.
[57] C. S. Henry, J. F. Zinner, M. P. Cohoon, and R. L. Stevens, “iBsu1103: A new genome-scale metabolic model of Bacillus subtilis based on SEED annotations,” Genome Biology, vol. 10, no. 6, pp. 1–15, 2009.
[58] R. S. Malik-Sheriff, M. Glont, T. V. Nguyen, K. Tiwari, M. G. Roberts, A. Xavier, M. T. Vu, J. Men, M. Maire, S. Kananathan, E. L. Fairbanks, J. P. Meyer, C. Arankalle, T. M. Varusai, V. Knight-Schrijver, L. Li, C. Dueñas-Roca, G. Dass, S. M. Keating, Y. M. Park, N. Buso, N. Rodriguez, M. Hucka, and H. Hermjakob, “BioModels-15 years of sharing computational models in life
science,” Nucleic Acids Research, vol. 48, no. D1, pp. D407–D415, 2020.
[59] L. Heirendt, S. Arreckx, T. Pfau, S. N. Mendoza, A. Richelle, A. Heinken, H. S. Haraldsdóttir, J. Wachowiak, S. M. Keating, V. Vlasov, S. Magnusdóttir, C. Y. Ng, G. Preciat, A. Žagare, S. H. Chan, M. K. Aurich, C. M. Clancy, J. Modamio, J. T. Sauls, A. Noronha, A. Bordbar, B. Cousins, D. C. El Assal, L. V. Valcarcel, I. Apaolaza, S. Ghaderi, M. Ahookhosh, M. Ben Guebila, A. Kostromins, N. Sompairac, H. M. Le, D. Ma, Y. Sun, L. Wang, J. T. Yurkovich, M. A.
Oliveira, P. T. Vuong, L. P. El Assal, I. Kuperstein, A. Zinovyev, H. S. Hinton, W. A. Bryant, F. J. Aragón Artacho, F. J. Planes, E. Stalidzans, A. Maass, S. Vempala, M. Hucka, M. A. Saunders, C. D. Maranas, N. E. Lewis, T. Sauter, B. Palsson, I. Thiele, and R. M. Fleming, “Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0,” Nature Protocols,
vol. 14, no. 3, pp. 639–702, 2019.
[60] Z. A. King, A. Dräger, A. Ebrahim, N. Sonnenschein, N. E. Lewis, and B. O. Palsson, “Escher: A Web Application for Building, Sharing, and Embedding Data-Rich Visualizations of Biological Pathways,” PLoS Computational Biology, vol. 11, no. 8, pp. 1–13, 2015.
[61] E. Fischer and U. Sauer, “Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism,” Nature Genetics, vol. 37, no. 6, pp. 636–640, 2005.
[62] BD, “BD Bionutrients TM Technical Manual.” 2015.
[63] S. H. Gorissen, J. J. Crombag, J. M. Senden, W. A. Waterval, J. Bierau, L. B. Verdijk, and L. J. van Loon, “Protein content and amino acid composition of commercially available plant-based protein isolates,” Amino Acids, vol. 50, no. 12, pp. 1685–1695, 2018.
[64] M. Dauner, T. Storni, and U. Sauer, “Bacillus subtilis metabolism and energetics in carbon-limited and excess-carbon chemostat culture,” Journal of Bacteriology, vol. 183, no. 24, pp. 7308–7317, 2001.
[65] P. D. Karp, R. Billington, R. Caspi, C. A. Fulcher, M. Latendresse, A. Kothari, I. M. Keseler, M. Krummenacker, P. E. Midford, Q. Ong, W. K. Ong, S. M. Paley, and P. Subhraveti, “The BioCyc collection of microbial genomes and metabolic pathways,” Briefings in Bioinformatics, vol. 20, no. 4, pp. 1085–1093, 2018.
[66] F. Fisher, “polyparci.” MATLAB Central File Exchange, https://www.mathworks.com/matlabcentral/fileexchange/39126-polyparci, 2014.
[67] Engineering Toolbox, “Air - Composition and Molecular Weight.” https://www.engineeringtoolbox.com/air-composition-d_212.html, 2003.
[68] J. Schellenberger, J. O. Park, T. M. Conrad, and B. Ø. Palsson, “BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions,” BMC Bioinformatics, vol. 11, no. 1, p. 213, 2010.
[69] F. Kunst, N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessières, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S.-K. Choi, J.-J. Codani, I. F. Connerton, N. J. Cummings, R. A. Daniel, F. Denizot, K. M. Devine, A. Düsterhöft, S. D. Ehrlich, P. T. Emmerson, K. D. Entian, J. Errington, C. Fabret, E. Ferrari, D. Foulger, C. Fritz, M. Fujita, Y. Fujita, S. Fuma, A. Galizzi, N. Galleron, S.-Y. Ghim, P. Glaser, A. Goffeau, E. J. Golightly, G. Grandi, G. Guiseppi, B. J. Guy, K. Haga, J. Haiech, C. R. Harwood, A. Hénaut, H. Hilbert, S. Holsappel, S. Hosono, M.-F. Hullo, M. Itaya, L. Jones, B. Joris, D. Karamata, Y. Kasahara, M. Klaerr-Blanchard, C. Klein, Y. Kobayashi, P. Koetter, G. Koningstein, S. Krogh, M. Kumano, K. Kurita, A. Lapidus, S. Lardinois, J. Lauber,
V. Lazarevic, S.-M. Lee, A. Levine, H. Liu, S. Masuda, C. Mauël, C. Médigue, N. Medina, R. P. Mellado, M. Mizuno, D. Moestl, S. Nakai, M. Noback, D. Noone, M. O’Reilly, K. Ogawa, A. Ogiwara, B. Oudega, S.-H. Park, V. Parro, T. M. Pohl, D. Portetelle, S. Porwollik, A. M. Prescott, E. Presecan, P. Pujic, B. Purnelle, G. Rapoport, M. Rey, S. Reynolds, M. Rieger, C. Rivolta, E. Rocha, B. Roche, M. Rose, Y. Sadaie, T. Sato, E. Scanlan, S. Schleich, R. Schroeter,
F. Scoffone, J. Sekiguchi, A. Sekowska, S. J. Seror, P. Serror, B.-S. Shin, B. Soldo, A. Sorokin, E. Tacconi, T. Takagi, H. Takahashi, K. Takemaru, M. Takeuchi, A. Tamakoshi, T. Tanaka, P. Terpstra, A. Tognoni, V. Tosato, S. Uchiyama, M. Vandenbol, F. Vannier, A. Vassarotti, A. Viari, R. Wambutt, E. Wedler, H. Wedler, T. Weitzenegger, P. Winters, A. Wipat, H. Yamamoto, K. Yamane,
K. Yasumoto, K. Yata, K. Yoshida, H.-F. Yoshikawa, E. Zumstein, H. Yoshikawa, and A. Danchin, “The complete genome sequence of the Gram-positive bacterium Bacillus subtilis,” Nature, vol. 390, pp. 249–256, nov 1997.
[70] S. Freeman, Biological Science. Prentice Hall, 2 ed., 2005.
[71] G. P. Rédei, Encyclopedia of genetics, genomics, proteomics, and informatics. Springer Science & Business Media, 2008.
[72] Y. Soma, K. Tsuruno, M. Wada, A. Yokota, and T. Hanai, “Metabolic flux redirection from a central metabolic pathway toward a synthetic pathway using a metabolic toggle switch,” Metabolic Engineering, vol. 23, pp. 175–184, 2014.
[73] K. G. Gadkar, F. J. Doyle, J. S. Edwards, and R. Mahadevan, “Estimating optimal profiles of genetic alterations using constraint-based models,” Biotechnology and Bioengineering, vol. 89, no. 2, pp. 243–251, 2005.
[74] M. Kanehisa, “KEGG: Kyoto Encyclopedia of Genes and Genomes,” Nucleic Acids Research, vol. 28, pp. 27–30, jan 2000.
[75] C. Stephens, “Bacterial sporulation: A question of commitment?,” Current Biology, vol. 8, no. 2, pp. 45–48, 1998.
[76] P. Pharkya, A. P. Burgard, and C. D. Maranas, “OptStrain: A computational framework for redesign of microbial production systems,” Genome Research, vol. 14, no. 11, pp. 2367–2376, 2004.
[77] W. Malfait, Analyse & calculus. Leuven: Acco, 2016.








