
Artificiële intelligentie kan kankercellijnen identificeren en hun biologisch karakteristieke kenmerken onderstrepen
Kankers zijn de afgelopen tien jaar veel beter behandelbaar. Dat heeft alles te maken met de opmars van therapieën die toegespitst zijn op de patiënt, een behandelingsfilosofie die de naam ‘persoonlijke geneeskunde’ draagt. Hierbij vraagt men zich eerst af waar het biologisch fout gaat in kankercellen, waarna men een therapie kiest die het best deze fouten uitbuit. Hierbij is het van cruciaal belang om kanker types te onderscheiden op basis van biologische kenmerken. Dit is een taak waar artificiële intelligentie een grote impact kan hebben. Zo hebben wij een AI-model geleerd om succesvol kankertypes van cellen te identificeren. Bovendien laten onze analyses toe om de onderlinge gelijkenissen en verschillen van kankertypes in kaart te brengen. Deze resultaten kunnen mogelijks leiden tot snellere diagnoses van kankers en nieuwe therapeutische strategieën.
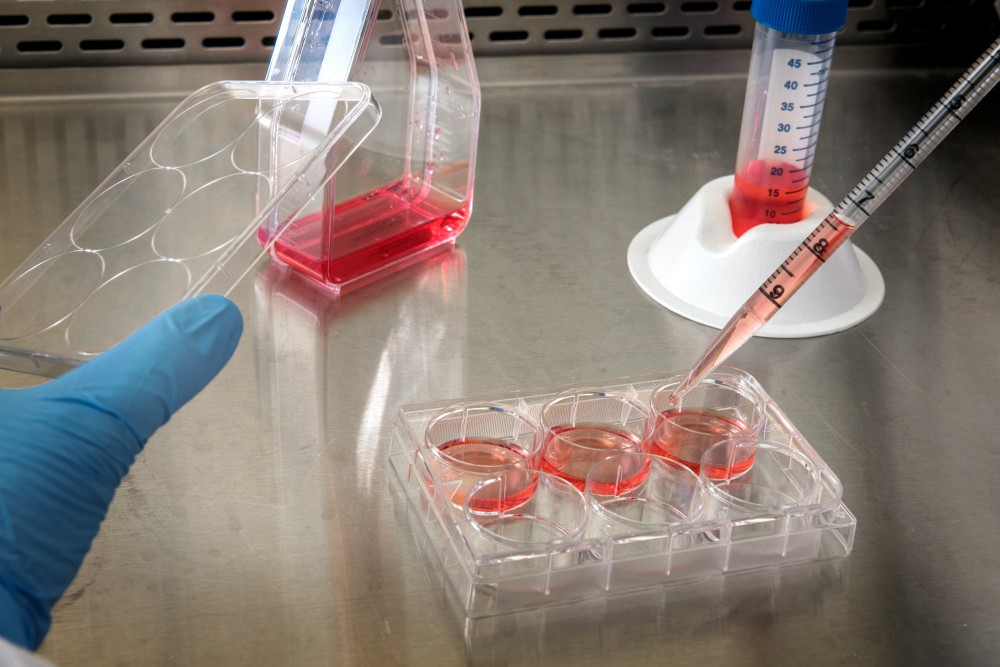
Cellijnen zijn cruciaal in kankeronderzoek
Het doel van een medicijn is om kankercellen van patiënten te doden. Helaas is het onmogelijk om honderdduizenden medicijnen direct op mensen te testen. Daarom maken onderzoekers gebruik van kankercellijnen. Dit zijn kankercellen afkomstig van patiënten die onsterfelijk zijn gemaakt en kunnen overleven in een plastic schaaltje. Deze technologie maakt het onderzoekers over heel de wereld mogelijk om met dezelfde soort kanker te werken en experimenten te herhalen. Zo wordt er al meer dan 70 jaar gewerkt met een kankercellijn afkomstig van Henrietta Lacks, een Afro-Amerikaanse vrouw die in 1951 overleed aan baarmoederhalskanker.
Onderzoekers werken al meer dan 70 jaar met een kankercellijn afkomstig van een vrouw die in 1951 overleed aan baarmoederhalskanker.
Ondertussen zijn er duizenden cellijnen ontwikkeld die verschillende types kanker modelleren. Kankertypes verschillen van elkaar door de type cel dat aanleiding geeft tot kanker of door welke mechanismen er abnormaal werken in de cel. Op basis van deze eigenschappen, is een bepaald type medicijn het meest effectief. Daarom is gedetailleerd identificeren en classificeren van kankercellen een van de hoekstenen van de persoonlijke geneeskunde. Om dit mogelijk te maken, zoeken onderzoekers naar moleculaire kenmerken die enkel aanwezig zijn in bepaalde kankers en de kankers onderling kunnen onderscheiden. Hierbij kijkt men vooral naar specifieke mutaties of een abnormale hoeveelheid of activiteit van eiwitten in de kankercellen.
Data-recyclage helpt AI om cellijnen te herkennen
Kankercellijnen zijn al meerdere decennia het favoriete biologisch modelsysteem van de onderzoeker. Maar pas sinds kort analyseren onderzoeksteams cellijnen op ongeziene schaal: de eiwitsamenstelling van honderden tot wel duizenden cellijnen worden geanalyseerd in grote, internationale studies. Deze berg van data is opgeslagen in databanken die elke geïnteresseerde onderzoeker en zelfs burger gratis kan raadplegen. Door deze vrije toegankelijkheid is het mogelijk om nieuwe conclusies te trekken uit reeds gepubliceerde data.
Zo hebben wij publieke databanken gebruikt om een AI-model aan te leren kankercellijnen te herkennen. Het AI-model baseert zich hierbij alleen maar op de proteïnesamenstelling van de kankercellijn. Door het model te vragen hoe het een kankercellijn herkent, kunnen onderzoekers inzicht krijgen in de meest kenmerkende proteïnen van de kankercellijn. Deze ‘sleuteleiwitten’ kunnen als aanvangspunt dienen om een kankertype beter te begrijpen. Bovendien kunnen dokters deze eiwitten mogelijks gebruiken om sneller kanker vast te stellen en vroeger een gepersonaliseerde therapie op te starten.
Cel-identiteit ontmaskerd! … of toch niet?
Wanneer onderzoekers een biologisch proces willen bestuderen, kiest men een gepaste cellijn om de experimenten op uit te voeren. Doordat andere onderzoekers dezelfde cellijn kunnen aankopen, kunnen deze experimenten opnieuw uitgevoerd en gevalideerd worden. Deze reproduceerbaarheid is een cruciale eigenschap van de wetenschap. Maar soms loopt het mis. Wanneer onderzoekers de cellijnen bewaren, kunnen deze overgenomen worden door een andere cellijn die beter groeit. Als gevolg daarvan werken de onderzoekers onvermoed met een andere cellijn, wat de resultaten en conclusies kan beïnvloeden.
Dit probleem kan verholpen worden met behulp van ons AI-model. Het model schat in hoe sterk de onbekende cellen lijken op alle voorheen gepubliceerde en bruikbare cellijndata. Cel-identificatie was voorheen ook mogelijk met DNA-methoden, waarbij de ‘DNA-barcode’ van de cellijnen werd gebruikt. In tegenstelling tot deze DNA-methoden, focust ons model op de bioactieve bestanddelen van de cel, met name de eiwitten. Kankercellen gedragen zich als kameleons, ze passen zich buitengewoon goed aan hun omgeving aan. Daarom is het belangrijk om niet alleen naar de identiteit van de cel te kijken, maar ook naar hoe de cel zich gedraagt in zijn plastic kweekschaaltje. Dit wil echter ook zeggen dat twee cellen van eenzelfde cellijn sterk verschillend kunnen reageren op een medicijn! Iets waar ons model geen rekening mee houdt.
Kankercellen gedragen zich als kameleons, ze passen zich buitengewoon goed aan hun omgeving aan
Uit ons onderzoek bleek dat niet alleen het kameleongedrag van kankercellen de reproduceerbaarheid van de wetenschap kan beïnvloeden. Ook verschillen in experimentele procedures zorgen voor onvergelijkbare resultaten. Dit is niet alleen te verklaren door de verschillende voeding van de cellen, maar ook over hoe de eiwitten uit de cellen worden geïsoleerd. Omdat het AI-model hier geen rekening mee hield tijdens het leerproces, kunnen hierdoor foutjes optreden in de identiteitsvoorspellingen van onbekende stalen.
Een naam is niet genoeg
Hoe kunnen we cellen classificeren wanneer hun namen geen nauwkeurige weergave bieden? Voor deze uitdaging kunnen recentere en geavanceerdere AI-technieken een oplossing bieden. Door alle gepubliceerde eiwitprofielen van cellijnen te combineren, kan een neuraal netwerkmodel de cel beschrijven met behulp van een beperkt aantal abstracte eigenschappen. Dit schept een meer genuanceerd beeld over de identiteit van de cel. Bovendien kan het model de kankercellen van een patiënt op een vergelijkbare manier beschrijven en aangeven welke cellijn het meest overeenkomt met de kanker van de patiënt. Zo kunnen cellijnexperimenten informatie bieden over veelbelovende therapeutische opties op een gepersonaliseerde manier.
Naar een kankervrije wereld
De persoonlijke geneeskunde is een veelbelovende behandelingsfilosofie die de genezingskans van patiënten sterk kan verbeteren. Maar deze vorm van medicijnontwikkeling presenteert ook unieke uitdagingen. Zo is het belangrijk om het kanker type zo nauwkeurig mogelijk te identificeren en therapieën hierop af te stemmen. Met behulp van datarecyclage en de ontwikkeling van nieuwe geavanceerde AI-systemen kunnen we deze uitdagingen aangaan en een stap dichter zetten naar een kankervrije wereld.
Bibliografie
-
1 Falzone, L., Salomone, S. & Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front Pharmacol 9, 1300, doi:10.3389/fphar.2018.01300 (2018).
-
2 Liu, J., Dang, H. & Wang, X. W. The significance of intertumor and intratumor heterogeneity in liver cancer. Experimental & Molecular Medicine 50, e416-e416, doi:10.1038/emm.2017.165 (2018).
-
3 Turashvili, G. & Brogi, E. Tumor Heterogeneity in Breast Cancer. Front Med (Lausanne) 4, 227,
doi:10.3389/fmed.2017.00227 (2017).
-
4 Mirabelli, P., Coppola, L. & Salvatore, M. Cancer Cell Lines Are Useful Model Systems for Medical
Research. Cancers 11 (2019).
-
5 Rijal, G. & Li, W. Native-mimicking in vitro microenvironment: an elusive and seductive future for
tumor modeling and tissue engineering. Journal of Biological Engineering 12, 20,
doi:10.1186/s13036-018-0114-7 (2018).
-
6 Hanahan, D. & Weinberg, Robert A. Hallmarks of Cancer: The Next Generation. Cell 144, 646-674,
doi:https://doi.org/10.1016/j.cell.2011.02.013 (2011).
-
7 Arora, M. Cell culture media: a review. Mater methods 3, 24 (2013).
-
8 Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature
569, 503-508, doi:10.1038/s41586-019-1186-3 (2019).
-
9 Guo, T. et al. Quantitative Proteome Landscape of the NCI-60 Cancer Cell Lines. iScience 21, 664-
680, doi:10.1016/j.isci.2019.10.059 (2019).
-
10 Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer
drug sensitivity. Nature 483, 603-607, doi:10.1038/nature11003 (2012).
-
11 Domcke, S., Sinha, R., Levine, D. A., Sander, C. & Schultz, N. Evaluating cell lines as tumour
models by comparison of genomic profiles. Nature Communications 4, 2126,
doi:10.1038/ncomms3126 (2013).
-
12 Ben-David, U. et al. Genetic and transcriptional evolution alters cancer cell line drug response.
Nature 560, 325-330, doi:10.1038/s41586-018-0409-3 (2018).
-
13 Gisselsson, D., Lindgren, D., Mengelbier, L. H., Øra, I. & Yeger, H. Genetic bottlenecks and the
hazardous game of population reduction in cell line based research. Experimental Cell Research
316, 3379-3386, doi:https://doi.org/10.1016/j.yexcr.2010.07.010 (2010).
-
14 Yang, X., Wen, Y., Song, X., He, S. & Bo, X. Exploring the classification of cancer cell lines from
multiple omic views. PeerJ 8, e9440, doi:10.7717/peerj.9440 (2020).
-
15 Sud, A., Kinnersley, B. & Houlston, R. S. Genome-wide association studies of cancer: current
insights and future perspectives. Nat Rev Cancer 17, 692-704, doi:10.1038/nrc.2017.82 (2017).
-
16 Tam, V. et al. Benefits and limitations of genome-wide association studies. Nature Reviews
Genetics 20, 467-484, doi:10.1038/s41576-019-0127-1 (2019).
-
17 Dugger, S. A., Platt, A. & Goldstein, D. B. Drug development in the era of precision medicine. Nat
Rev Drug Discov 17, 183-196, doi:10.1038/nrd.2017.226 (2018).
-
18 Boyle, E. A., Li, Y. I. & Pritchard, J. K. An Expanded View of Complex Traits: From Polygenic to
Omnigenic. Cell 169, 1177-1186, doi:10.1016/j.cell.2017.05.038 (2017).
-
19 Correa Rojo, A. et al. Towards Building a Quantitative Proteomics Toolbox in Precision Medicine: A
Mini-Review. Frontiers in Physiology 12 (2021).
-
20 Liu, Y., Beyer, A. & Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA
Abundance. Cell 165, 535-550, doi:https://doi.org/10.1016/j.cell.2016.03.014 (2016).
-
21 Nusinow, D. P. et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387-
402.e316, doi:https://doi.org/10.1016/j.cell.2019.12.023 (2020).
-
22 Gonçalves, E. et al. Pan-cancer proteomic map of 949 human cell lines. Cancer Cell 40, 835-
849.e838, doi:10.1016/j.ccell.2022.06.010 (2022).
-
23 Gholami, Amin M. et al. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Reports 4,
609-620, doi:https://doi.org/10.1016/j.celrep.2013.07.018 (2013).
-
24 Jarnuczak, A. F. et al. An integrated landscape of protein expression in human cancer. Scientific
Data 8, 115, doi:10.1038/s41597-021-00890-2 (2021).
-
25 Kustatscher, G. et al. Co-regulation map of the human proteome enables identification of protein
functions. Nat Biotechnol 37, 1361-1371, doi:10.1038/s41587-019-0298-5 (2019).
![]()
![]()
![]()
![]()
![]()
-
26 Schwarz, A. & Beck, M. The Benefits of Cotranslational Assembly: A Structural Perspective. Trends Cell Biol 29, 791-803, doi:10.1016/j.tcb.2019.07.006 (2019).
-
27 Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry- based proteomics evidences. Nucleic Acids Research 50, D543-D552, doi:10.1093/nar/gkab1038 (2022).
-
28 Claeys, T., Menu, M., Bouwmeester, R., Gevaert, K. & Martens, L. Machine Learning on Large- Scale Proteomics Data Identifies Tissue and Cell-Type Specific Proteins. Journal of Proteome Research, doi:10.1021/acs.jproteome.2c00644 (2023).
-
29 Duong, V.-A. & Lee, H. Bottom-Up Proteomics: Advancements in Sample Preparation. International Journal of Molecular Sciences 24 (2023).
-
30 Glatter, T., Ahrné, E. & Schmidt, A. Comparison of Different Sample Preparation Protocols Reveals Lysis Buffer-Specific Extraction Biases in Gram-Negative Bacteria and Human Cells. Journal of Proteome Research 14, 4472-4485, doi:10.1021/acs.jproteome.5b00654 (2015).
-
31 Varnavides, G. et al. In Search of a Universal Method: A Comparative Survey of Bottom-Up Proteomics Sample Preparation Methods. J Proteome Res 21, 2397-2411, doi:10.1021/acs.jproteome.2c00265 (2022).
-
32 Feist, P. & Hummon, A. B. Proteomic challenges: sample preparation techniques for microgram- quantity protein analysis from biological samples. Int J Mol Sci 16, 3537-3563, doi:10.3390/ijms16023537 (2015).
-
33 Rozanova, S. et al. in Quantitative Methods in Proteomics (eds Katrin Marcus, Martin Eisenacher, & Barbara Sitek) 85-116 (Springer US, 2021).
-
34 Davies, V. et al. Rapid Development of Improved Data-Dependent Acquisition Strategies. Analytical Chemistry 93, 5676-5683, doi:10.1021/acs.analchem.0c03895 (2021).
-
35 Kreimer, S. et al. Advanced Precursor Ion Selection Algorithms for Increased Depth of Bottom-Up Proteomic Profiling. J Proteome Res 15, 3563-3573, doi:10.1021/acs.jproteome.6b00312 (2016).
-
36 Lin, T.-T. et al. Mass spectrometry-based targeted proteomics for analysis of protein mutations. Mass Spectrometry Reviews 42, e21741, doi:https://doi.org/10.1002/mas.21741 (2023).
-
37 Verheggen, K. et al. Anatomy and evolution of database search engines—a central component of mass spectrometry based proteomic workflows. Mass Spectrometry Reviews 39, 292-306, doi:https://doi.org/10.1002/mas.21543 (2020).
-
38 The UniProt, C. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research 51, D523-D531, doi:10.1093/nar/gkac1052 (2023).
-
39 Degroeve, S. et al. ionbot: a novel, innovative and sensitive machine learning approach to LC- MS/MS peptide identification. bioRxiv, 2021.2007.2002.450686, doi:10.1101/2021.07.02.450686 (2022).
-
40 Dupree, E. J. et al. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 8, doi:10.3390/proteomes8030014 (2020).
-
41 Bouwmeester, R., Gabriels, R., Hulstaert, N., Martens, L. & Degroeve, S. DeepLC can predict retention times for peptides that carry as-yet unseen modifications. Nat Methods 18, 1363-1369, doi:10.1038/s41592-021-01301-5 (2021).
-
42 Declercq, A. et al. Updated MS2PIP web server supports cutting-edge proteomics applications. Nucleic Acids Research, gkad335, doi:10.1093/nar/gkad335 (2023).
-
43 Ahrné, E., Molzahn, L., Glatter, T. & Schmidt, A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. PROTEOMICS 13, 2567-2578, doi:https://doi.org/10.1002/pmic.201300135 (2013).
-
44 Brenes, A., Hukelmann, J., Bensaddek, D. & Lamond, A. I. Multibatch TMT Reveals False Positives, Batch Effects and Missing Values. Mol Cell Proteomics 18, 1967-1980, doi:10.1074/mcp.RA119.001472 (2019).
-
45 Florens, L. et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40, 303-311, doi:10.1016/j.ymeth.2006.07.028 (2006).
-
46 Wiśniewski, J. R., Hein, M. Y., Cox, J. & Mann, M. A "proteomic ruler" for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics 13, 3497-3506, doi:10.1074/mcp.M113.037309 (2014).
![]()
![]()
![]()
-
47 Bubis, J. A., Levitsky, L. I., Ivanov, M. V., Tarasova, I. A. & Gorshkov, M. V. Comparative evaluation of label-free quantification methods for shotgun proteomics. Rapid Communications in Mass Spectrometry 31, 606-612, doi:https://doi.org/10.1002/rcm.7829 (2017).
-
48 Chawade, A., Alexandersson, E. & Levander, F. Normalyzer: A Tool for Rapid Evaluation of Normalization Methods for Omics Data Sets. Journal of Proteome Research 13, 3114-3120, doi:10.1021/pr401264n (2014).
-
49 Callister, S. J. et al. Normalization Approaches for Removing Systematic Biases Associated with Mass Spectrometry and Label-Free Proteomics. Journal of Proteome Research 5, 277-286, doi:10.1021/pr050300l (2006).
-
50 Välikangas, T., Suomi, T. & Elo, L. L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Briefings in Bioinformatics 19, 1-11, doi:10.1093/bib/bbw095 (2018).
-
51 Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118-127, doi:10.1093/biostatistics/kxj037 (2007).
-
52 Lazar, C., Gatto, L., Ferro, M., Bruley, C. & Burger, T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. Journal of Proteome Research 15, 1116-1125, doi:10.1021/acs.jproteome.5b00981 (2016).
-
53 Desaire, H., Go, E. P. & Hua, D. Advances, obstacles, and opportunities for machine learning in proteomics. Cell Reports Physical Science 3, 101069, doi:https://doi.org/10.1016/j.xcrp.2022.101069 (2022).
-
54 Webb-Robertson, B.-J. M. et al. Review, Evaluation, and Discussion of the Challenges of Missing Value Imputation for Mass Spectrometry-Based Label-Free Global Proteomics. Journal of Proteome Research 14, 1993-2001, doi:10.1021/pr501138h (2015).
-
55 McCoy, J. T., Kroon, S. & Auret, L. Variational Autoencoders for Missing Data Imputation with Application to a Simulated Milling Circuit. IFAC-PapersOnLine 51, 141-146, doi:https://doi.org/10.1016/j.ifacol.2018.09.406 (2018).
-
56 Lualdi, M. & Fasano, M. in Proteomics Data Analysis (ed Daniela Cecconi) 143-159 (Springer US, 2021).
-
57 Aboudi, N. E. & Benhlima, L. in 2016 International Conference on Engineering & MIS (ICEMIS). 1- 5.
-
58 Shi, Z., Wen, B., Gao, Q. & Zhang, B. Feature Selection Methods for Protein Biomarker Discovery from Proteomics or Multiomics Data. Molecular & Cellular Proteomics 20, 100083, doi:https://doi.org/10.1016/j.mcpro.2021.100083 (2021).
-
59 Nanga, S. et al. Review of dimension reduction methods. J. Data Anal. Inf. Process. 09, 189-231, doi:10.4236/jdaip.2021.93013 (2021).
-
60 Sharma, S., Gosain, A. & Jain, S. in International Conference on Innovative Computing and Communications. (eds Ashish Khanna et al.) 459-472 (Springer Singapore).
-
61 Kovács, G. An empirical comparison and evaluation of minority oversampling techniques on a large number of imbalanced datasets. Applied Soft Computing 83, 105662, doi:https://doi.org/10.1016/j.asoc.2019.105662 (2019).
-
62 Chawla, N. V., Bowyer, K. W., Hall, L. O. & Kegelmeyer, W. P. SMOTE: Synthetic Minority over- Sampling Technique. J. Artif. Int. Res. 16, 321–357 (2002).
-
63 Fernández, A., Garcia, S., Herrera, F. & Chawla, N. SMOTE for Learning from Imbalanced Data: Progress and Challenges, Marking the 15-year Anniversary. Journal of Artificial Intelligence Research 61, 863-905, doi:10.1613/jair.1.11192 (2018).
-
64 Islam, Z., Abdel-Aty, M., Cai, Q. & Yuan, J. Crash data augmentation using variational autoencoder. Accident Analysis & Prevention 151, 105950, doi:https://doi.org/10.1016/j.aap.2020.105950 (2021).
-
65 Asperti, A. & Trentin, M. Balancing Reconstruction Error and Kullback-Leibler Divergence in Variational Autoencoders. IEEE Access 8, 199440-199448, doi:10.1109/ACCESS.2020.3034828 (2020).
-
66 Way, G. P. & Greene, C. S. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. Pac Symp Biocomput 23, 80-91 (2018).
![]()
![]()
![]()
![]()
![]()
![]()
-
67 Lundberg, S. M. & Lee, S.-I. in Advances in Neural Information Processing Systems Vol. 30 (eds I. Guyon et al.) (Curran Associates, Inc., 2017).
-
68 Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J Biomol Tech 29, 25-38, doi:10.7171/jbt.18-2902-002 (2018).
-
69 The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49, D325-d334, doi:10.1093/nar/gkaa1113 (2021).
-
70 Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419, doi:10.1126/science.1260419 (2015).
-
71 Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 51, D638-d646, doi:10.1093/nar/gkac1000 (2023).
-
72 Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology 157, 105-132, doi:https://doi.org/10.1016/0022-2836(82)90515-0 (1982).
-
73 Abdelkader, B., Julien, H., Chloé-Agathe, A. & Akpéli, N. pyComBat, a Python tool for batch effects correction in high-throughput molecular data using empirical Bayes methods. bioRxiv, 2020.2003.2017.995431, doi:10.1101/2020.03.17.995431 (2021).
-
74 Slany, A. et al. Contribution of Human Fibroblasts and Endothelial Cells to the Hallmarks of Inflammation as Determined by Proteome Profiling. Molecular & cellular proteomics : MCP 15, 1982-1997, doi:10.1074/mcp.m116.058099 (2016).
-
75 Nakada, M. et al. Integrin α3 is overexpressed in glioma stem-like cells and promotes invasion. Br J Cancer 108, 2516-2524, doi:10.1038/bjc.2013.218 (2013).
-
76 Toyoda, H., Nagai, Y., Kojima, A. & Kinoshita-Toyoda, A. Podocalyxin as a major pluripotent marker and novel keratan sulfate proteoglycan in human embryonic and induced pluripotent stem cells. Glycoconjugate Journal 34, 817-823, doi:10.1007/s10719-017-9801-8 (2017).
-
77 Bernal, A. & Arranz, L. Nestin-expressing progenitor cells: function, identity and therapeutic implications. Cell Mol Life Sci 75, 2177-2195, doi:10.1007/s00018-018-2794-z (2018).
-
78 Patel, V. J. et al. A Comparison of Labeling and Label-Free Mass Spectrometry-Based Proteomics Approaches. Journal of Proteome Research 8, 3752-3759, doi:10.1021/pr900080y (2009).
-
79 Saltzman, A. B. et al. gpGrouper: A Peptide Grouping Algorithm for Gene-Centric Inference and Quantitation of Bottom-Up Proteomics Data. Mol Cell Proteomics 17, 2270-2283, doi:10.1074/mcp.TIR118.000850 (2018).
-
80 Claeys, T. et al. (Research Square Platform LLC, 2023).
-
81 Choksawangkarn, W., Edwards, N., Wang, Y., Gutierrez, P. & Fenselau, C. Comparative study of
workflows optimized for in-gel, in-solution, and on-filter proteolysis in the analysis of plasma
membrane proteins. J Proteome Res 11, 3030-3034, doi:10.1021/pr300188b (2012).
-
82 Tiwari, P., Kaila, P. & Guptasarma, P. Understanding anomalous mobility of proteins on SDS-
PAGE with special reference to the highly acidic extracellular domains of human E- and N-
cadherins. Electrophoresis 40, 1273-1281, doi:10.1002/elps.201800219 (2019).
-
83 Asperti, A. in Machine Learning, Optimization, and Data Science. (eds Giuseppe Nicosia et al.)
297-308 (Springer International Publishing).










