Gevangen in je eigen lichaam.
Gevangen in je eigen lichaam.
Beeld je in dat je niet meer kan bewegen. Je ligt volledig stil op je bed en kan enkel nog rond je kijken. Praten gaat moeizaam. Ademen ga at moeizaam. Je hebt jeuk maar je kan er niet bij. Je hebt dorst maar je kan het glas water niet grijpen. Je zit gevangen in een lichaam vol pijn. Wanneer je gediagnosticeerd wordt met Amyotrofe Laterale Sclerose, afgekort als ALS, is dit het vooruitzicht. Een doeltreffende behandeling is er tot op vandaag niet. ALS hebben is meer dan een doodsvonnis. Het is een lijdensweg.
at moeizaam. Je hebt jeuk maar je kan er niet bij. Je hebt dorst maar je kan het glas water niet grijpen. Je zit gevangen in een lichaam vol pijn. Wanneer je gediagnosticeerd wordt met Amyotrofe Laterale Sclerose, afgekort als ALS, is dit het vooruitzicht. Een doeltreffende behandeling is er tot op vandaag niet. ALS hebben is meer dan een doodsvonnis. Het is een lijdensweg.
Wereldwijd worden elke dag 328 mensen gediagnosticeerd met ALS. De ziekte treft zowel mannen als vrouwen, met een gemiddeld leeftijd van 55 jaar oud maar er zijn ook tieners met ALS. Deze patiënten ondervinden krachtverlies en worden vervolgens geleidelijk aan verlamd. De oorzaak is het verlies van zenuwcellen die instaan voor de motoriek (motorneuronen genoemd). Deze cellen zorgen voor de signaaloverdracht tussen de hersenen en de spieren. Zonder motorneuronen zijn je spieren nog intact en heb je wel de wil om te bewegen, maar het commando zal jouw spieren niet bereiken (Fig. 1). Alles wat je bent en kan sijpelt langzaamaan weg. Het merendeel van de ALS-patiënten sterft binnen twee tot vijf jaar doordat ook de ademhalingspieren verlammen. Kortom, ze stikken. Op dit moment is er geen eenduidige hypothese die het verlies aan motorneuronen volledig kan verklaren. Dit bemoeilijkt de ontwikkeling van een doeltreffend geneesmiddel. Daarom is wetenschappelijk onderzoek naar waarom en hoe deze motorneuronen afsterven noodzakelijk.
Zal ontsnapping ooit mogelijk zijn?
Zenuwcellen van ALS-patiënten zijn enkel in een beperkte hoeveelheid beschikbaar. Daarom worden er dierenmodellen, zoals vliegen, vissen en muizen gebruikt voor wetenschappelijk onderzoek. Jarenlang onderzoek met deze modellen heeft onze kennis over ALS vergroot maar heeft tot op vandaag niet kunnen leiden tot een geneesmiddel die de ziekte stopt. Eén van de oorzaken is dat ALS-patiënten en ziektemodellen niet identiek zijn. Ten eerste, dieren verschillen van mensen. Bovendien is de complexiteit van de ontwikkeling van deze ziekte in mensen niet volledig aanwezig. In deze dierenmodellen ontwikkeld ALS zich artificieel door het aanbrengen van een genetisch wijziging (een mutatie) in slechts één gen. Dit is in overeenkomst met een kleine groep van ALS-patiënten maar is verschillend van de oorzaak van ALS in de meeste patiënten. Er wordt verondersteld dat in deze groep een complexe combinatie van meerdere genen en omgevingsfactoren een rol speelt in het ziekteproces.
Het gebruik van geïnduceerde pluripotente stamcellen (iPSCn) biedt ons hier een oplossing aan. Hiervoor vertrekken we vanuit de huidcellen van ALS-patiënten (Fig.2). Het voordeel is dat huidcellen op een eenvoudige manier kunnen verkregen worden in tegenstelling tot zenuwcellen. Vervolgens worden deze huidcellen terug getransformeerd naar een “basiscel”, namelijk stamcellen. Deze stamcellen gekend als iPSCn hebben de mogelijkheid om, wanneer blootgesteld aan de juiste factoren, elke soort cel in het menselijke lichaam te vormen. In onze studie werd aangetoond dat deze stamcellen succesvol kunnen worden omgezet in motorneuronen, de cellen die afsterven bij ALS. Dit is in overeenkomst met andere studies en heeft ons de mogelijkheid om te werken met menselijke weefsels, in de plaats van dierenmodellen.
Figuur 1. Bron: the Inspiration room; ALS-campagne “No signal in Canada”. Fotograaf Russell Monk en Ben Weeks.
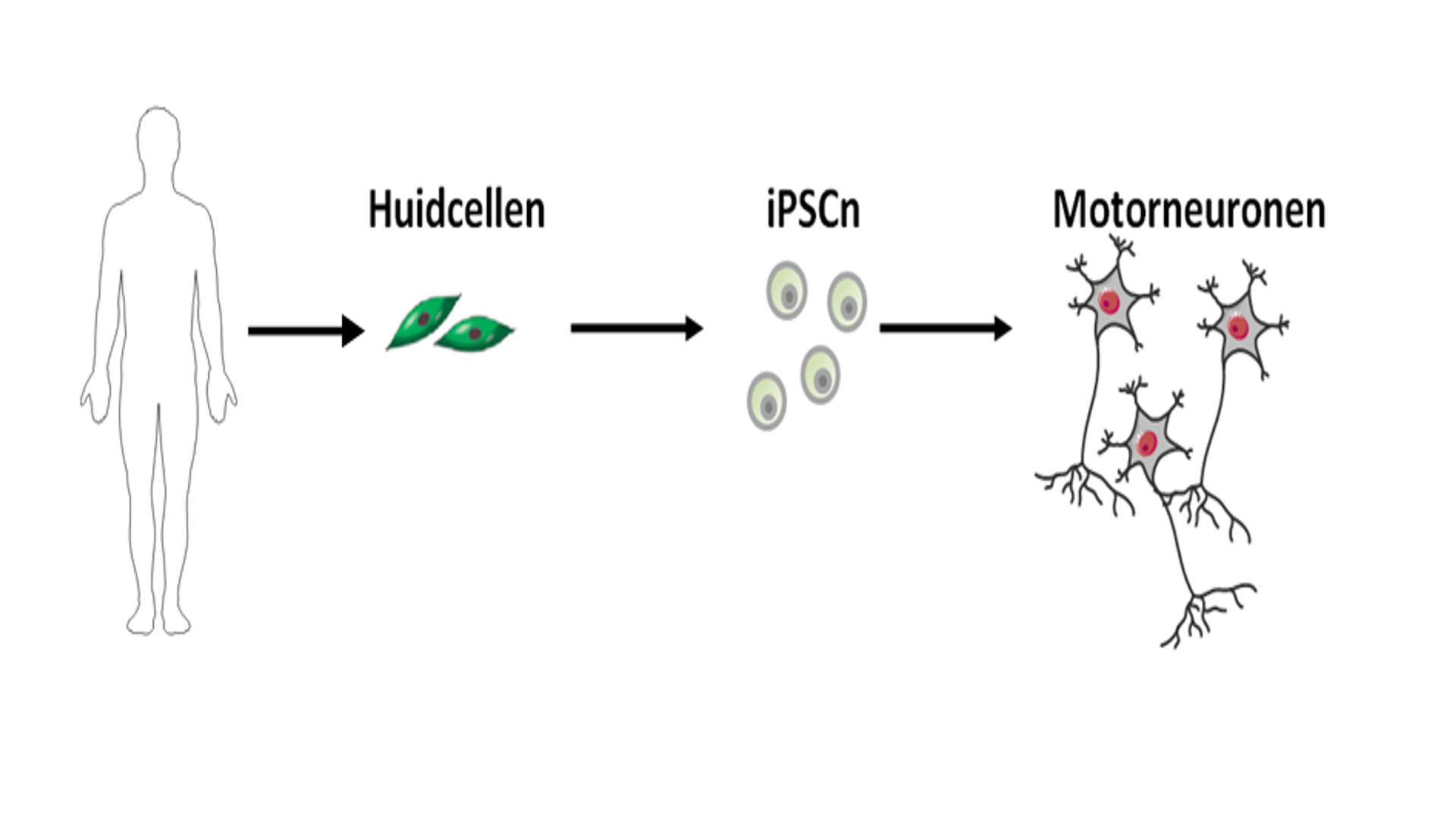
Figuur 2. De huidcellen van ALS-patiënten worden gebruikt om via een tussenstap van geïnduceerde pluripotente stamcellen (iPSCn) motorneuronen te ontwikkelen. Motorneuronen zijn de belangrijkste cellen in het ALS-onderzoek omdat deze cellen afsterven bij ALS-patiënten. Motorneuronen rechtstreek verkrijgen van de patiënten is een schadelijke en niet efficiënte methode. Bron figuren: medical servier.
Maar er is meer! Er werden verschillen opgemerkt tussen motorneuronen afkomstig van ALS-patiënten en motorneuronen afkomstig van gezonde personen. Een motorneuron bestaat uit een klein cellichaam en een erg lange uitloper, die het axon wordt genoemd (Fig. 3). Het cellichaam is het “hart” van het neuron waar nieuwe elementen worden aangemaakt en afgebroken. Het kan worden opgedeeld in de kern en het omliggende cytoplasma (Fig. 3). Het axon zorgt voor de verbinding tussen de hersenen (waar het cellichaam ligt) en de spieren. Een axon kan in het menselijke lichaam meer dan 1 meter lang zijn! Daarom is transport langs dit axon essentieel voor een goede gezondheid van het motorneuron. Goede elementen worden zo verdeeld over het lange axon en beschadigde elementen worden afgevoerd naar het cellichaam voor hun afbraak. Onze studies wezen erop dat er een verstoring is van dit axonaal transport. Naast dit verstoord transport werd ook een mislocalisatie waargenomen van een bekend ALS-geassocieerd eiwit, namelijk FUS (Fused in Sarcoma). FUS is normaal sterk aanwezig in de kern van het cellichaam. Echter, in de ALS-cellijnen werd een hogere aanwezigheid van FUS waargenomen in het cytoplasma (Fig. 4), wat een negatief effect kan hebben op het motorneuron.
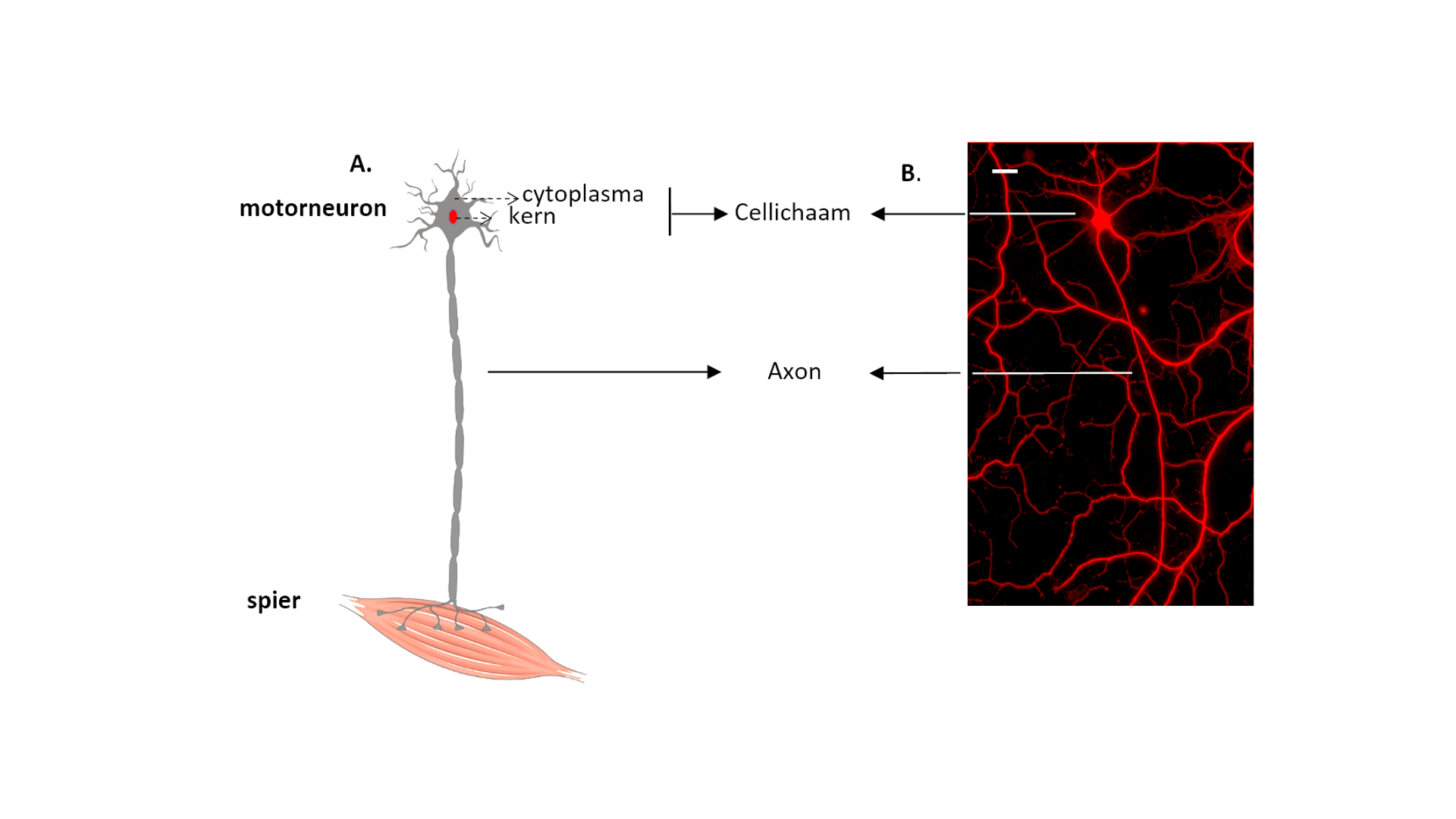
Figuur 3. Het motorneuron. (A) Een schematische weergave van een motorneuron en (B) een met fluorescent gemerkte motorneuron verkregen vanuit iPSCn. (A) Bron figuur: medical servier (B) Schaalbar= 20 µm.
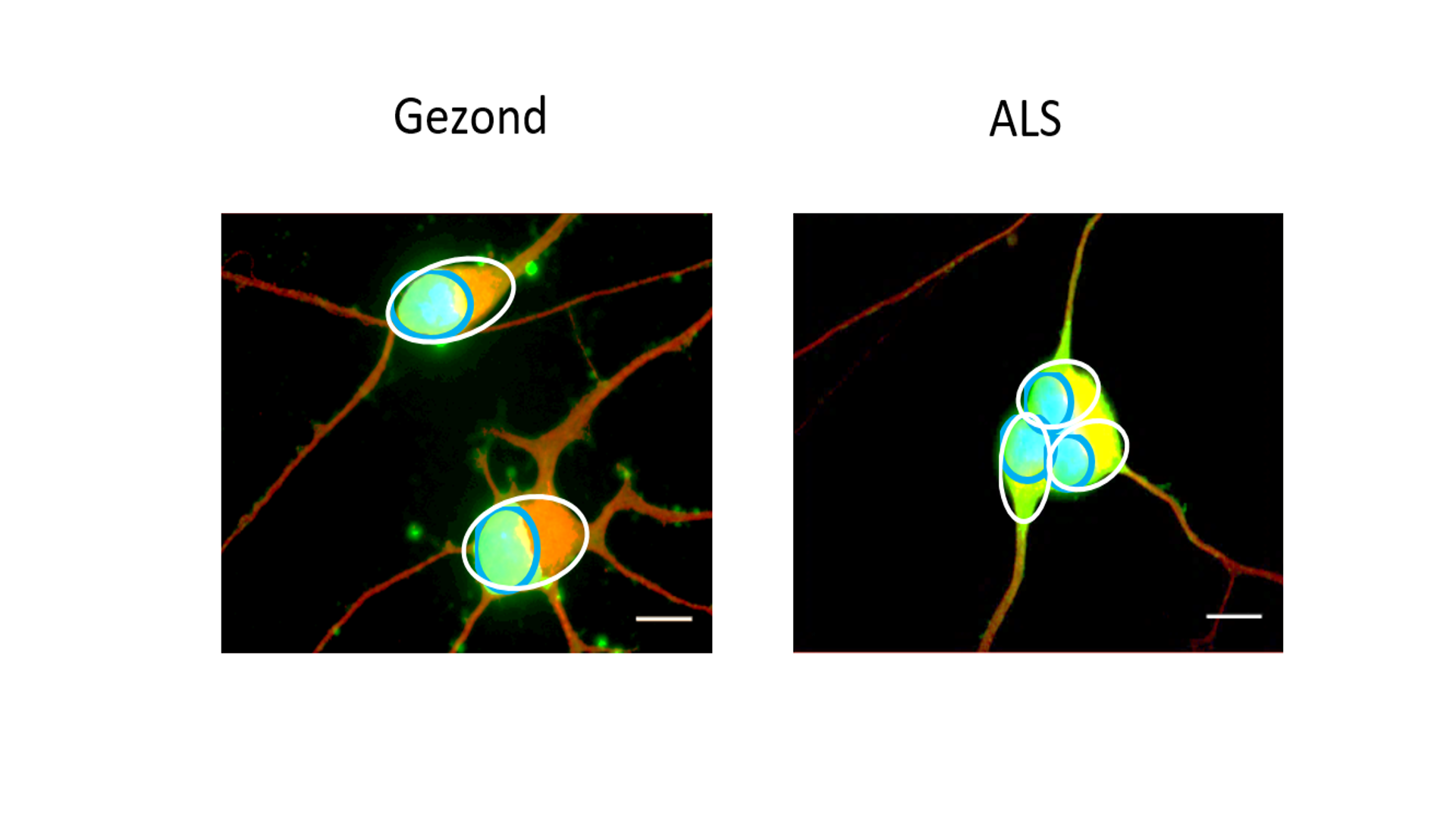
Figuur 4. FUS mislocalisatie. iPSC-afgeleide motorneuronen van een gezonde persoon en een ALS-patiënt gemerkt met fluorescente merkers. Rood = het motorneuron. Blauw= de kern; omgeven door een blauwe cirkel. Het cytoplasma is omgeven door een witte cirkel. Groen= FUS. Geel= overlap tussen het cytoplasma en FUS. Merk op dat FUS meer gelocaliseerd is in het cytoplasma (gele kleur) bij de motorneuronen afgeleid van de ALS-patiënt. Schaalbar= 40 µm.
Veel werk moet er nog worden verzet voordat wetenschappers een duidelijk beeld zullen hebben over wat er precies fout loopt. Hoopvol is dat de opkomst van iPSCn een nieuwe wind brengt door het ALS-onderzoek. Deze nieuwe inzichten leiden dan misschien wel tot de ontwikkeling van een geneesmiddel die de ziekte stopt of zelfs geneest, zodat de mensen getroffen door ALS niet langer gevangen zullen zitten in hun eigen lichaam.
Bibliografie
References
1. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. Nature Publishing Group; 2013;14(4):248–64.
2. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3.
3. Rowland LP, Shneider NA. Amyotrophic Lateral Sclerosis. new Engl J. 2001;344(22):1688–700.
4. Iguchi Y, Katsuno M, Ikenaka K, Ishigaki S, Sobue G. Amyotrophic lateral sclerosis: An update on recent genetic insights. J Neurol. 2013;260(11):2917–27.
5. Laferriere F, Polymenidou M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med Wkly. 2015;(January):1–13.
6. Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. Nature Publishing Group; 2014;17(1):17–23.
7. Gros-Louis F, Gaspar C, Rouleau G a. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11-12):956–72.
8. Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. Nature Publishing Group; 2013;9(11):617–28.
9. Alonso a., Logroscino G, Jick SS, Hernán M a. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol. 2009;16(6):745–51.
10. Mancuso R, Navarro X. Amyotrophic lateral sclerosis: Current perspectives from basic research to the clinic. Prog Neurobiol. Elsevier Ltd; 2015 Aug 5;26.
11. Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–23.
12. Bensimon G, Lacomblez L, Meninger V, Group AS. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330(9):585–91.
13. Richard J-P, Maragakis NJ. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Res. Elsevier; 2014;1607:15–25.
14. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;363:210–1.
15. Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta - Mol Basis Dis. Elsevier B.V.; 2015;1852(4):679–84.
16. Mackenzie IR a., Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–34.
17. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. 2009;323(February):1208–11.
18. Buratti E, Baralle F. Multiple Roles of TDP-43 in gene expression, splicing regulation and human disease. Front Biosci. 2008;13:867–78.
19. Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136(6):1001–4.
20. Yang S, Warraich ST, Nicholson G a., Blair IP. Fused in sarcoma/translocated in liposarcoma: A multifunctional DNA/RNA binding protein. Int J Biochem Cell Biol. Elsevier Ltd; 2010;42(9):1408–11.
21. Lissouba A, Liao M, Brustein E, Guy A, Kabashi E. FUS and TARDBP but Not SOD1 Interact in Genetic Models of Amyotrophic Lateral Sclerosis. 2011;7(8):17–28.
22. Baloh RH. How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr Opin Neurol. 2012;25(6):1.
23. Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. Nature Publishing Group; 2012;15(11):1488–97.
24. Szafranski K. Non-coding RNA in neural function, disease, and aging. Front Genet. 2015;6(March):1–16.
25. King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. Elsevier B.V.; 2012;1462:61–80.
26. Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. Nature Publishing Group; 2014;10(6):337–48.
27. Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. Elsevier Ltd; 2012;11(4):297–8.
28. Levine TP, Daniels RD, Gatta a. T, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29(4):499–503.
29. Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EMC, Parkinson G, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1–6.
30. Lee Y-B, Chen H-J, Peres JN, Gomez-Deza J, Attig J, Štalekar M, et al. Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep. 2013;5(5):1178–86.
31. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in als and FTD: Disrupted RNA and protein homeostasis. Neuron. Elsevier Inc.; 2013;79(3):416–38.
32. Cozzolino M, Ferri A, Carrì MT. Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal. 2008;10(3):405–43.
33. Perry TL, Krieger C, Hansen S, Eisen A. Amyotrophic lateral sclerosis: amino acid levels in plasma and cerebrospinal fluid. Ann Neurol. 1990;28(1):12–7.
34. Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta - Mol Basis Dis. 2006;1762(11-12):1068–82.
35. Doble A. The Role of Excitotoxicity in Neurodegenerative Disease: implications for therapy. Pharmacol Ther. 1999;81(3):163–221.
36. Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34(4-5):325–37.
37. Bellingham MC. A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis: What have we Learned in the Last Decade? CNS Neurosci Ther. 2011;17(1):4–31.
38. Mitsumoto H, Santella R., Liu X, Bodganov M, Zipprich J, Wu H-C, et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph lateral Scler. 2008;9:177–83.
39. Nicholls DG, Budd SL. Mitochondria and Neuronal Survival. Physiol Rev. 2000;80(1):315–60.
40. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39(1):359–407.
41. Cozzolino M, Carrì MT. Mitochondrial dysfunction in ALS. Prog Neurobiol. 2012;97(2):54–66.
42. Sasaki S, Maruyama S, Yamane K, Sakuma H, Takeishi M. Ultrastructure of swollen proximal axons of anterior horn neurons in motor neuron disease. J Neurol Sci. 1990;97(2-3):233–40.
43. Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66(1):10–6.
44. Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18(9):3241–50.
45. Cai Q, Tammineni P. Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front Cell Neurosci. 2016;10(February):1–17.
46. Millecamps S, Julien J-P. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. Nature Publishing Group; 2013;14(3):161–76.
47. Chevalier-Larsen E, Holzbaur ELF. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762(11-12):1094–108.
48. Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43(1):19–30.
49. Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29(16):2841–57.
50. Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43 splicing of an intron within the 3′ untranslated region of its own transcript, thereby triggering nonsense mediated RNA degradation. (147 words). Nat Neurosci. Nature Publishing Group; 2011;14(4):459–68.
51. Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011;63(September):n/a – n/a.
52. Atkin JD, Farg M a., Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. Elsevier Inc.; 2008;30(3):400–7.
53. Atkin JD, Farg M a., Turner BJ, Tomas D, Lysaght J a., Nunan J, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281(40):30152–65.
54. Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. 2013;8(4):888–99.
55. Fischer LR, Culver DG, Tennant P, Davis A a., Wang M, Castellano-Sanchez A, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–40.
56. Cavanagh, J. B. The significance of the “dying back” process in human and experimental neurological diseases. Int Rev Exp. 1964;3:219–67.
57. Spencer, P S. Central-peripheral distal axonopathy - The pathogenisis of dying-back polyneuropathies. Prog Neuropathol. 1976;3:253–95.
58. Holzbaur ELF. Motor neurons rely on motor proteins. Trends Cell Biol. 2004;14(5):233–40.
59. Perlson E, Maday S, Fu M-M, Moughamian AJ, Holzbaur ELF. Retrograde axonal transport: pathways to cell death? Trends Neurosci. Elsevier Ltd; 2010;33(7):335–44.
60. Vos KJ De, Grierson AJ, Ackerley S, Miller CCJ. Role of Axonal Transport in Neurodegenerative Diseases. Annu Rev Neurosci. 2008;31:151–73.
61. Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995. p. 61–4.
62. Perlson E, Jeong G-B, Ross JL, Dixit R, Wallace KE, Kalb RG, et al. A Switch in Retrograde Signaling from Survival to Stress in Rapid-Onset Neurodegeneration. J Neurosci. 2009;29(31):9903–17.
63. Ligon L a, LaMonte BH, Wallace KE, Weber N, Kalb RG, Holzbaur ELF. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport. 2005;16(6):533–6.
64. Bommel H, Xie G, Rossoll W, Wiese S. Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J Cell Biol. 2002;159(4):563–9.
65. Sagot Y, Vejsada R, Kato AC, Sagot Y, Vejsada R, Kato AC. Clinical and molecular aspects of motoneurone diseases : animal models , neurotrophic factors and Bcl-2 oncoprotein. Trends Pharmacol. 1997;18:330–7.
66. Ferri A, Sanes, J R, Coleman, M P, Cunningham, J M, Kato, A C. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motorneuron disease. Curr Biol. 2003;13:669–73.
67. Ikenaka K, Katsuno M, Kawai K, Ishigaki S, Tanaka F, Sobue G. Disruption of axonal transport in motor neuron diseases. Int J Mol Sci. 2012;13(1):1225–38.
68. Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300(5620):808–12.
69. LaMonte BH, Wallace KE, Holloway B a., Shelly SS, Ascaño J, Tokito M, et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34(5):715–27.
70. Humbert, Richard; David A. Adler, Christine M. Disteche, Christopher Hassett, Curtis J. Omiecinski CEF. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation (12;16) in malignant liposarcoma. Nat Genet. 1993;3:73–96.
71. Prasad DD, Ouchida M, Lee L, Rao VN, Reddy ES. TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t(16;21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene. 1994;9(12):3717–29.
72. Tan AY, Manley JL. The TET family of proteins: Functions and roles in disease. J Mol Cell Biol. 2009;1(2):82–92.
73. Peters OM, Ghasemi M, Brown RH. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125(5):1767–79.
74. Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110 ( Pt 1:1741–50.
75. Sama RRK, Ward CL, Bosco D a. Functions of FUS/TLS From DNA Repair to Stress Response: Implications for ALS. ASN Neuro. 2014;6(4).
76. Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J. 2000;19(3):453–62.
77. Zhu H, Belcher M, van der Harst P. Healthy aging and disease: role for telomere biology? Clin Sci (Lond). 2011;120(10):427–40.
78. Meissner M, Lopato S, Gotzmann J, Sauermann G, Barta A. Proto-oncoprotein TLS/FUS is associated to the nuclear matrix and complexed with splicing factors PTB, SRm160, and SR proteins. Exp Cell Res. 2003;283(2):184–95.
79. Orozco D, Edbauer D. FUS-mediated alternative splicing in the nervous system: Consequences for ALS and FTLD. J Mol Med. 2013;91(12):1343–54.
80. Morlando M, Dini Modigliani S, Torrelli G, Rosa A, Di Carlo V, Caffarelli E, et al. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. Nature Publishing Group; 2012;31(24):4502–10.
81. Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, et al. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15(6):587–93.
82. Fiesel FC, Kahle PJ. TDP-43 and FUS/TLS: Cellular functions and implications for neurodegeneration. FEBS J. 2011;278(19):3550–68.
83. Dormann D, Haass C. Fused in sarcoma (FUS): An oncogene goes awry in neurodegeneration. Mol Cell Neurosci. Elsevier Inc.; 2013;56:475–86.
84. Hübers A, Just W, Rosenbohm A, Müller K, Marroquin N, Goebel I, et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging. Elsevier Inc; 2015;36(11):3117.e1–3117.e6.
85. Mackenzie IR a, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. Elsevier Ltd; 2010;9(10):995–1007.
86. Zhang ZC, Chook YM. Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc Natl Acad Sci. 2012;109(30):12017–21.
87. Kent L, Vizard TN, Smith BN, Topp SD, Vance C, Gkazi A, et al. Autosomal dominant inheritance of rapidly progressive amyotrophic lateral sclerosis due to a truncation mutation in the fused in sarcoma (FUS) gene. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7-8):557–62.
88. Sama RRK, Ward CL, Kaushansky LJ, Lemay N, Ishigaki S, Urano F, et al. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J Cell Physiol. 2013;228(11):2222–31.
89. Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33(3):141–50.
90. Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. Elsevier Inc.; 2013;154(4):727–36.
91. Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. Elsevier Inc.; 2015;162(5):1066–77.
92. Schaefer A, O’Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, et al. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med. 2007;204(7):1553–8.
93. Tao J, Wu H, Lin Q, Wei W, Lu X-H, Cantle JP, et al. Deletion of Astroglial Dicer Causes Non-Cell-Autonomous Neuronal Dysfunction and Degeneration. J Neurosci. 2011;31(22):8306–19.
94. Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem. 2014;289(2):1192–202.
95. Yang L, Gal J, Chen J, Zhu H. Self-assembled FUS binds active chromatin and regulates gene transcription. PNAS. 2014;111(50):17809–14.
96. Julien J-P, Kriz J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11-12):1013–24.
97. Benatar M. Lost in translation: Treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis. 2007;26(1):1–13.
98. McGoldrick P, Joyce PI, Fisher EMC, Greensmith L. Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta. Elsevier B.V.; 2013;1832(9):1421–36.
99. Dunckley T, Huentelman MJ, Craig DW, Pearson J V, Szelinger S, Joshipura K, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med. 2007;357(8):775–88.
100. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131(5):861–72.
101. Davis-Dusenbery BN, Williams L a, Klim JR, Eggan K. How to make spinal motor neurons. Development. 2014;141(3):491–501.
102. Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science (80- ). 2008;321(5893):1218–21.
103. Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat Neurosci. 2016;19(4).
104. Hamburger V. The Heritage of Experimental Embryology: Hans Spemann and the Organizer. oxford Univ Press. 1988;
105. Stern CD. Neural induction: old problem, new findings, yet more questions. Development. 2005;132(9):2007–21.
106. Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, et al. Drug Screening for ALS Using Patient-Specific Induced Pluripotent Stem Cells. Sci Transl Med. 2012;4(145):145ra104–45ra104.
107. Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 2012;109(15):5803–8.
108. Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. Elsevier Inc.; 2013;56:355–64.
109. Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–99.
110. Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry N a., Vidensky S, et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron. Elsevier; 2013;80(2):415–28.
111. Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell. Elsevier Inc.; 2014;14(6):796–809.
112. Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281(5384):1851–4.
113. Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Steve SW, Berry JD, et al. Intrinsic membrane hyperexcitability of ALS patient-derived motor neurons. Cell Rep. 2014;7(1):1–11.
114. Japtok J, Lojewksi X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J, et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis. Elsevier Inc.; 2015;82:420–9.
115. Lenzi J, De Santis R, de Turris V, Molando M, Laneve P, Calvo A, et al. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis Model Mech. 2015;8(7):755–66.
116. Matus S, Medinas DB, Hetz C. Common ground: Stem cell approaches find shared pathways underlying ALS. Cell Stem Cell. Elsevier Inc.; 2014;14(6):697–9.
117. Maury Y, Côme J, Piskorowski R a, Salah-Mohellibi N, Chevaleyre V, Peschanski M, et al. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol. 2014;33(1):89–96.
118. Sheng Z-H, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13(2):77–93.
119. Davis-Dusenbery BN, Williams L a, Klim JR, Eggan K. How to make spinal motor neurons. Development. 2014;141(3):491–501.
120. Amoroso MW, Croft GF, Williams DJ, O’Keeffe S, Carrasco M a, Davis a R, et al. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci. 2013;33(2):574–86.
121. Sareen D, O’Rourke JG, Meera P, Muhammad a KMG, Grant S, Simpkinson M, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149.
122. Devlin A-C, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, et al. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. Nature Publishing Group; 2015;6:1–12.
123. Qu Q, Li D, Louis KR, Li X, Yang H, Sun Q, et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat Commun. Nature Publishing Group; 2014;5:3449.
124. Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. HHS Public Access. Cell Stem Cell. 2015;512(7512):49–53.
125. Sun Y, Dykes IM, Liang X, Eng SR, Evans SM, Turner EE. A central role for Islet1 in sensory neuron development linking sensory and spinal gene regulatory programs. Nat Neurosci. 2008;11(11):1283–93.
126. Wilson JM, Hartley R, Maxwell DJ, Todd AJ, Lieberam I, Kaltschmidt JA, et al. Conditional Rhythmicity of Ventral Spinal Interneurons Defined by Expression of the Hb9 Homeodomain Protein. J Neurosci. 2005;25(24):5710–9.
127. Liu X, Chen J, Liu W, Li X, Chen Q, Liu T, et al. The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics. 2015;16(3):223–31.
128. Ichiyanagi N, Fujimori K, Yano M, Ishihara-Fujisaki C, Sone T, Akiyama T, et al. Establishment of In Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Reports. The Authors; 2016;6(4):496–510.
129. Kaus A, Sareen D. ALS Patient Stem Cells for Unveiling Disease Signatures of Motoneuron Susceptibility: Perspectives on the Deadly Mitochondria, ER Stress and Calcium Triad. Front Cell Neurosci. 2015;9(November):448.
130. Philippidou P, Dasen JS. Hox Genes: Choreographers in Neural Development, Architects of Circuit Organization. Neuron. Elsevier Inc.; 2013;80(1):12–34.
131. Zou ZY, Cui LY, Sun Q, Li XG, Liu MS, Xu Y, et al. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol Aging. Elsevier Ltd; 2013;34(4):1312.e1–1312.e8.
132. Huang EJ, Zhang J, Geser F, Trojanowski JQ, Strober JB, Dickson DW, et al. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. brain pathol. 2010;20(6):1069–76.
133. Leblond CS, Webber a., Gan-Or Z, Moore F, Dagher a., Dion P a., et al. De novo FUS P525L mutation in Juvenile amyotrophic lateral sclerosis with dysphonia and diplopia. Neurol Genet. 2016;2(2):e63–e63.
134. Shang Y, Huang EJ. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. Elsevier; 2016;1–14.
135. Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22(13):2676–88.
136. Murakami T, Yang SP, Xie L, Kawano T, Fu D, Mukai A, et al. Als mutations in FUS cause neuronal dysfunction and death in caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet. 2012;21(1):1–9.
137. Duffy LM, Chapman a. L, Shaw PJ, Grierson a. J. The role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2011;37(4):336–52.
138. Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ. Axonal transport defects are a common phenotype in Drosophila models of ALS. oxford Univ Press. 2016;
139. Lee S, Sato Y, Nixon R a. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31(21):7817–30.
140. Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur ELF. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron. Elsevier Inc.; 2014;84(2):292–309.
141. Maday S, Wallace KE, Holzbaur ELF. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196(4):407–17.
142. Yang Y, Coleman M, Zhang L, Zheng X, Yue Z. Autophagy in axonal and dendritic degeneration. Trends Neurosci. Elsevier Ltd; 2013;36(7):418–428.
143. Wong YC, Holzbaur ELF. Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci. 2015;128(7):1259–67.
144. Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. Nature Publishing Group; 2011;8(2):108–17.
145. Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron. Elsevier Inc.; 2015;87(2):355–71.
146. Groen EJN, Fumoto K, Blokhuis AM, Engelen-Lee JY, Zhou Y, van den Heuvel DM a, et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet. 2013;22(18):3690–704.
147. Gerbino V, Carrì MT, Cozzolino M, Achsel T. Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol Dis. Elsevier Inc.; 2013;55:120–8.
148. Garcera a, Bahi N, Periyakaruppiah a, Arumugam S, Soler RM. Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell Death Dis. Nature Publishing Group; 2013;4(6):e686.
149. Xu C, Denton KR, Wang Z, Zhang X, Li X. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Model Mech. 2016;9:39–49.
150. Pierzynska-Mach A, Janowski P a., Dobrucki JW. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytom Part A. 2014;85(8):729–37.
151. Cai Q, Lu L, Tian J, Zhu Y, Qiao H, Sheng Z. Snapin-Regulated Late Endosomal Transport Is Critical for Efficient Autophagy-Lysosomal Function in Neurons. Neuron. 2010;68:73–86.
152. Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, et al. Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell. Elsevier Inc.; 2014;14(6):796–809.
153. Magrané J, Cortez C, Gan W-B, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23(6):1413–24.
154. Daigle GG, Lanson N a., Smith RB, Casci I, Maltare A, Monaghan J, et al. Rna-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet. 2013;22(6):1193–205.
155. Miller J, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, et al. Human iPSC-based modeling of late onset disease via progerin-induced aging. 2013. 691-705 p.
156. Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, et al. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem. 2012;287(19):15635–47.
157. Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi T a, et al. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. Nature Publishing Group; 2011;18(12):1428–31.
158. Edgar JM, Nave KA. The role of CNS glia in preserving axon function. Curr Opin Neurobiol. 2009;19(5):498–504.
159. Haidet-Phillips AM, Gross SK, Williams T, Tuteja A, Sherman A, Ko M, et al. Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp Neurol. Elsevier Inc.; 2013;250:250–9.
160. Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2013;110(12):4697–702.
161. Jacquier a., Bellouze S, Blanchard S, Bohl D, Haase G. Astrocytic protection of spinal motor neurons but not cortical neurons against loss of Als2/alsin function. Hum Mol Genet. 2009;18(12):2127–39.
162. Chandran J, Ding J, Cai H. Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol Neurobiol. 2007;36(3):224–31.








